All animal experiments were performed according to NIH recommendations contained within the Guide for the Care and Use of Laboratory Animals and were approved by the IACUC at BioLegend.
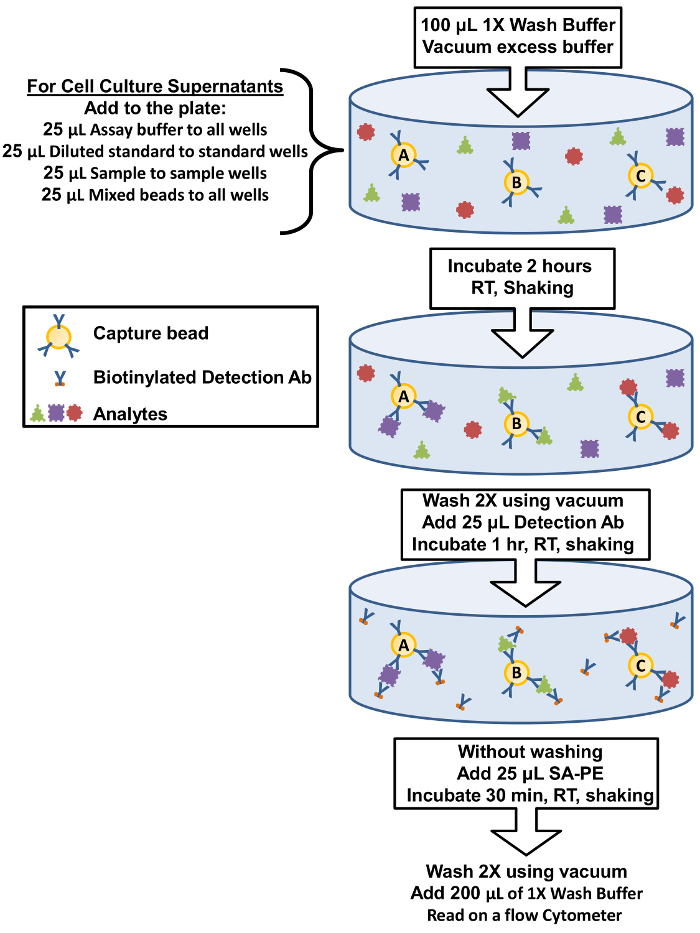
Figure 1: Filter Plate Assay Procedure Summary. The protocol for performing the assay using filter plates is represented as a diagram depicting key assay components and incubation steps. Please click here to view a larger version of this figure.
1. Biological Sample Preparation
NOTE: The chosen anatomical site and volume of blood collection is left to the discretion of each individual investigator and will not affect the outcome of the assay. However, once they are obtained, samples should be handled according to the steps listed below.
- Preparation of Serum Samples
- Collect the desired volume of blood into preferred collection tube and allow it to clot for at least 30 min.
- Centrifuge the sample for 10 min at 1,000 x g at room temperature.
- Remove serum and assay immediately or aliquot into polypropylene microcentrifuge tubes and store samples at ≤ -20 °C.
- Preparation of Plasma Samples
- Collect the desired volume of blood using ethylenediaminetetraacetic acid (EDTA) coated blood collection tubes.
- Centrifuge for 10 min at 1,000 x g at room temperature within 30 min of collection.
- Remove plasma and assay immediately, or aliquot into polypropylene microcentrifuge tubes and store samples at ≤ -20 °C.
- Preparation of Tissue Culture Supernatant Samples
- Centrifuge the sample for 10 min at 1,500 x g at 4 °C to remove cells and debris.
- Collect supernatant and assay immediately or aliquot into polypropylene microcentrifuge tubes and store at ≤ -20 °C.
- Thawing of Frozen Biological Samples
- Completely thaw any previously frozen biological samples on ice, and vortex briefly before use. Avoid more than 2 freeze/thaw cycles.
NOTE: The samples used in this protocol were obtained by culturing BALB/c mouse splenocytes (1 x 106 cells at 37 °C + 5% CO2 under various conditions: unstimulated, LPS (100 ng/mL), αCD3 (1 µg/mL plate-coated) + αCD28 (1 µg/mL soluble), PMA (20 ng/mL) + Ionomycin (500 ng/mL). Culture supernatants were collected after 48 h and processed as described in section 1.3.
- Completely thaw any previously frozen biological samples on ice, and vortex briefly before use. Avoid more than 2 freeze/thaw cycles.
2. Reagent Preparation
- Preparation of Pre-mixed Antibody-Immobilized Beads
- Sonicate pre-mixed beads bottle for 1 min in a sonicator bath at room temperature, and then vortex for 30 s prior to use. If no sonicator bath is available, increase the vortex time to 1 min.
- Preparation of Wash Buffer
- Bring the 20x Wash Buffer (20x PBS with 1% Tween-20) supplied with the kit to room temperature and vortex to bring all salts into solution.
- Dilute 25 mL of 20x Wash Buffer with 475 mL deionized water. Store unused portion between 2 °C and 8 °C for up to one month.
- Preparation of Matrix B (For Use with Serum and Plasma Samples Only)
- Add 5.0 mL of the Assay Buffer (PBS with 1% BSA) included in the kit to the bottle containing lyophilized Matrix B. Allow at least 15 min for complete reconstitution, then vortex to mix well. Leftover reconstituted Matrix B can be stored at ≤ -70 °C for up to one month.
3. Standard Preparation
NOTE: Each analyte in this panel has a top standard concentration of 10,000 pg/mL.
- Add 250 µL of Assay Buffer to reconstitute the lyophilized Mouse Th Cytokine Standard Cocktail. Mix by briefly vortexing and allow the vial to sit at room temperature for 10 min.
- Transfer the standard cocktail to a polypropylene microcentrifuge tube labeled "C7". This will be used as the top standard.
- Label 6 polypropylene microcentrifuge tubes as C6, C5, C4, C3, C2, and C1.
- Add 75 µL of Assay Buffer to each of these tubes.
- Transfer 25 µL of the top standard C7 to the C6 tube and mix well by vortexing. This will be the C6 standard.
- Continue performing serial 1:4 dilutions by using a new pipet tip for each tube to add 25 µL of the previous standard to the 75 µL of Assay Buffer in the next lowest standard tube followed by vortexing to obtain standards C5, C4, C3, C2, and C1. Use Assay Buffer as the 0 pg/mL standard (C0).
4. Sample Dilution
NOTE: Preliminary pilot experiments using this assay with multiple dilutions may be required to determine the most appropriate dilution factor for a particular set of biological samples. A proper dilution factor will produce concentration calculations for the sample that lie within the bounds of the standard curve. The following steps are meant as guidelines and may have to be determined empirically depending on sample type.
- Dilute serum or plasma samples 2-fold with Assay Buffer (e.g. dilute 50 µL of sample with 50 µL of Assay Buffer) in polypropylene microcentrifuge tubes.
NOTE: If further sample dilution is required, dilutions should be done with Matrix B instead of assay buffer to ensure accurate measurement. Adding serum or plasma samples without dilution will result in low assay accuracy and may clog the filter plate. Matrix B is comprised of pooled mouse serum depleted of endogenous assay targets. It is used as a serum or plasma sample diluent to avoid matrix effects, which are known to the affect analytical sensitivity of immunoassays. - Test cell culture supernatant samples without dilution.
NOTE: The levels of analyte can vary greatly from sample to sample. If necessary, dilute supernatant samples using a fresh preparation of their corresponding cell culture medium or Assay Buffer.
5. Assay Procedure
NOTE: The assay can be performed in polypropylene filter plates, micro FACS tubes, or V-bottom microplates. The filter plate assay procedure is recommended due to good sample to sample consistency, assay robustness, and ease of handling. This procedure requires a vacuum filtration unit for washing (provided by end-user).
- Allow all reagents to warm to room temperature (20 – 25 °C) before use.
- Set the filter plate on an inverted plate cover at all times during the assay setup and incubation steps, so that the bottom of the plate does not touch any surfaces as it may cause leaking.
- Keep the plate upright during the entire assay procedure, including the washing steps, to avoid losing beads.
- Keep the plate in the dark or wrapped with aluminum foil for all incubation steps.
- Run all standards and samples as duplicates, arranged on the plate in a sequential order.
- Pre-wet the filter plate by adding 100 µL of 1x Wash Buffer to each well and let it sit for 1 min at room temperature (if using filter-bottom microplate).
- Remove buffer volume by using the vacuum manifold (5 – 10 s). Do not exceed 10 " Hg of vacuum. Blot excess Wash Buffer from the bottom of the plate by pressing the plate on a stack of clean paper towels. Place the plate on top of the inverted plate cover.
NOTE: Steps 5.1 – 5.2 may be omitted if performing the assay using a V-bottom microplate or micro FACS tubes. - For cell culture supernatant samples, add 25 µL of Assay Buffer to all wells. Add 25 µL of each standard to the standard wells. Add 25 µL of each sample to the sample wells.
- For measuring serum or plasma samples, add 25 µL of Matrix B to the standard wells. Add 25 µL of Assay Buffer to the sample wells. Add 25 µL of each standard to the standard wells. Add 25 µL of each diluted serum or plasma sample to the sample wells.
- Vortex mixed beads for 30 s. Add 25 µL of mixed beads to each well, shaking bead bottle intermittently to avoid bead settling. The final volume should be 75 µL in each well after the addition of beads.
- Seal the plate with a plate sealer. Wrap the entire plate, including the inverted plate cover, with aluminum foil. Place the plate on a plate shaker, secure it, and shake at approximately 500 rpm for 2 h at room temperature. To prevent leaking, do not apply positive pressure to the plate sealer.
- Without inverting, place the plate on the vacuum manifold and apply vacuum as before and add 200 µL of 1x wash buffer to each well.
- Remove the contents of the assay plate wells by vacuum filtration. Blot excess wash buffer from the bottom of the plate with an absorbent pad or paper towels. Repeat this step one more time.
NOTE: If the assay is performed using a V-bottom microplate or micro FACS tubes skip steps 5.12 and 5.13. Instead centrifuge the plate at 1,000 x g for 5 min at room temperature, then remove the supernatant using a multichannel pipette. - Add 25 µL of detection antibodies (see the Table of Materials) to each well.
- Seal the plate with a fresh plate sealer. Wrap the entire plate, including the inverted plate cover, with aluminum foil.
- Place the plate on a plate shaker and shake at approximately 500 rpm for 1 h at room temperature.
- Without vacuuming, add 25 µL of SA-PE reagent directly to each well. No dilution of reagent is necessary.
- Seal the plate with a fresh plate sealer. Wrap the entire plate, including the inverted plate cover, with aluminum foil.
- Place the plate on a plate shaker and shake at approximately 500 rpm for 30 min at room temperature.
- Repeat step 5.13 above.
- Add 200 µL of 1x Wash Buffer to each well. Re-suspend the beads on a plate shaker for 1 min.
- Using a multichannel pipet, transfer samples from the filter plate to FACS tubes to read samples on a flow cytometer.
NOTE: Sample volume may be increased from 200 µL to 300 µL by adding an extra 100 µL of 1x Wash Buffer to each tube to avoid the sample running dry. If necessary samples may be stored at 4 °C protected from light and analyzed the following day. However, prolonged sample storage can lead to reduced signal.
6. Flow Cytometer Set-up
NOTE: In order to generate reliable data, the flow cytometer must be set up properly before data acquisition. This process will vary for individual users in some regards depending on the configuration of the particular instrument the assay is performed on. A generalized setup process for a cytometer able to measure PE and APC and equipped with both a 488 and 633 nm laser is discussed below.
- Start up the flow cytometer and acquisition software according to the manufacturer's instructions provided with the instrument.
- Create a dot plot with FSC (forward scatter) on the X-axis and SSC (side scatter) for the Y-axis. Set FSC and SSC to linear mode.
- Create a second dot plot with PE on the X-axis and APC for the Y-axis. This plot should be set to a log-based display mode.
- Vortex the vial of raw beads included in the kit for 30 s to re-suspend the beads. These beads contain an internal APC dye and are comprised of two size populations.
- Transfer 400 µL of the raw beads to a new FACS tube.
- Set the flow cytometer flow rate to low.
- Run the raw beads, carefully adjusting the gain and voltage for FSC and SSC so that both size populations of these beads are visibly separated and easy to gate.
- Adjust the FSC threshold to exclude unwanted events (i.e. debris or air bubbles).
- In the FSC vs. SSC plot, draw a gate that includes all bead populations.
NOTE: The raw beads contain two size populations of beads, the smaller "A beads" and the larger "B bead" regions. - Display the gated bead populations from the FSC vs. SSC plot on the second dot plot with PE on the X-axis and APC for the Y-axis.
- Adjust the PMT voltage for the APC fluorescence channel so that the APC signal for all bead populations has a median fluorescence intensity (MFI) that lies between 1 x 101 and 5 x 103.
- Vortex the vial of PE setup beads for 30 s to re-suspend the beads.
- Transfer 400 µL of the PE beads to a new FACS tube.
- Replace the raw beads tube from the flow cytometer with the PE beads tube.
- Adjust the photomultiplier tube (PMT) voltage for the PE fluorescence channel setting so that the MFI of the PE beads falls between the lot-specific range found listed on the PE beads vial.
NOTE: The PE setup beads contain beads of one population size ("A beads" only).
7. Data Acquisition
NOTE: The exact procedures associated with the acquisition of data on a given instrument can vary and are dependent on the cytometer's configuration specifications and the interface software used. The instructions below are therefore intended to highlight the required steps to be taken in the assay regardless of the cytometer employed in the assay.
- Verify the cytometer flow rate is still set to low.
- Set the number of bead events to be acquired to about 300 per analyte. For a 13-plex panel this equates to acquiring 3,900 events combined from both bead size populations (A+B beads).
- Vortex each sample for 5 s before analysis.
- Read samples. When reading samples, set the flow cytometer to setup mode first and wait until bead population is stabilized before switching to acquisition mode.
- Use simple names with consecutive numbering for data files to facilitate data analysis.
- Export only the gated events (A + B bead regions) rather than total events.
- Store all FCS files in the same folder for each assay. If running multiple assays, create a separate folder for each assay.
8. Proceed to Data Analysis
NOTE: The FCS files generated on the flow cytometer should be analyzed using the data analysis software, which can be downloaded for free14.
- Install the data analysis software on a PC.
- Transfer all assay FCS files to the computer that contains the analysis software.
- Plug the license key dongle (included in the kit) into a USB port of the computer.
- Launch the data analysis software.
- Click the blue "Add Files" button located at the top of the screen.
- Navigate to the folder that contains the FCS files from the assay in the pop up window that appears.
- Click and drag all assay FCS files from the pop up window to the software display. All files should now appear in a list.
- Click the green "Next" button on the bottom right of the display. Left-click and hold to drag the small blue standard curve buttons (C7 to C0) to their corresponding FCS files from the list to define the standard curve.
- Click the green "Next" button on the bottom right of the display to open the gating pop up window.
- On the left side of the gating window enter the names of both the A and B bead regions assay analyte targets and their associated bead ID (found in the manual supplied with the assay) in ascending order.
- Select the gating tool from the top of the pop up window and use it to draw 2 gates in the FSC vs. SSC plot, one around the A beads and a second around the B beads.
- Review the APC vs. PE scatter plots that appear below the FSC vs. SSC plot that appear. There will be 6 analyte bands in the A beads (left plot) and 7 analyte bands in the B beads (right plot).
NOTE: Gates may be redrawn manually by selecting the eraser tool at the top and clicking to delete a given gate. The gating tool may then be selected and used to manually apply a gate to the desired band region. - Click the green "OK" button to close the gating pop up window and return to the list of FCS files.
- Define any dilutions that were made to biological samples by right clicking a given file, selecting "Dilution Fold" from the menu, and input the correct value.
- Click the green "Run" button at the bottom right of the display to generate the standard curves for the assay and calculate the concentrations of unknown biological samples.
This protocol demonstrates how a bead-based immunoassay platform can be used to simultaneously quantitate cytokine profiles from biological samples using a flow cytometer. The biological samples used in this example were cell culture supernatants from mouse splenocytes that had been previously incubated under various activating conditions, although the assay format has also been validated for use with serum or plasma samples.
Data generated from the assay are used to construct standard curves for all analytes in the multiplexed panel. The data analysis software classifies each particular analyte based on a unique bead size and APC intensity, and quantifies them using a PE reporter. The dynamic ranges, as well as the sensitivity of the assay are demonstrated when plotted on a log-log scale in Figure 2A. The software uses assay data and a 5-parameter curve fitting algorithm to accurately calculate not only the concentrations of analytes in user provided biological samples but also the limits of detection on both the high and low end of all standard curves (Table 1). The format described in this protocol also provides highly robust assay precision. In Figure 2B and 2C data are presented depicting the intra-assay variability of each analyte. Two separate standard protein concentrations (high and low) were analyzed in one assay with 16 replicates for each sample. Figure 2C and 2D represent the inter-assay variability of target analytes from three independent assays. As with the intra-assay precision experiments, high and low standard concentrations were analyzed with 3 replicates in each of the independent experiments. Minimal variability in the calculated concentration of target proteins is clearly visible.
In order to demonstrate the utility of bead-based cytometric assays in interrogating cytokine expression profiles, mouse splenocytes were incubated under various activating conditions. In addition to an unstimulated control, cells were also incubated with LPS, anti-CD3/anti-CD28 antibodies, or phorbol 12-myristate 13-acetate and ionomycin. Cell culture supernatants were collected 48 h later and assayed using the Mouse T helper cytokine panel. The quantification results from this experiment are represented in Figure 3 and the effect of stimulation conditions on cytokine concentrations is clearly delineated.
The results described above are typical, as long as the experimenter has good laboratory technique and follows the provided assay protocol. Sources of error in this assay format can arise from deviations in incubation times, reagent volumes, washing, or from improper PMT settings on the flow cytometer used to analyze the samples. In Figure 4A a scatter plot from a successful assay shows 2 distinct bead populations that are clearly defined. This can be contrasted to Figure 4B, where poor washing and protocol adherence have led to bead aggregates which blur the two populations. In addition, as evidenced by the debris in lower left quadrant, total cytometer events were exported for analysis rather than the preferred format of gated events only. Proper cytometer set up is critical to generating reliable data in this assay. In Figure 4C proper PMT settings allow for the resolution of each of the 6 analytes in the APC classification channel. Improper PMT settings will result in data similar to that presented in Figure 4D where the APC channel has bead populations with overlapping classification intensities. This will invalidate the assay since it will be impossible to calculate analyte concentrations without clearly defined populations of assay beads.
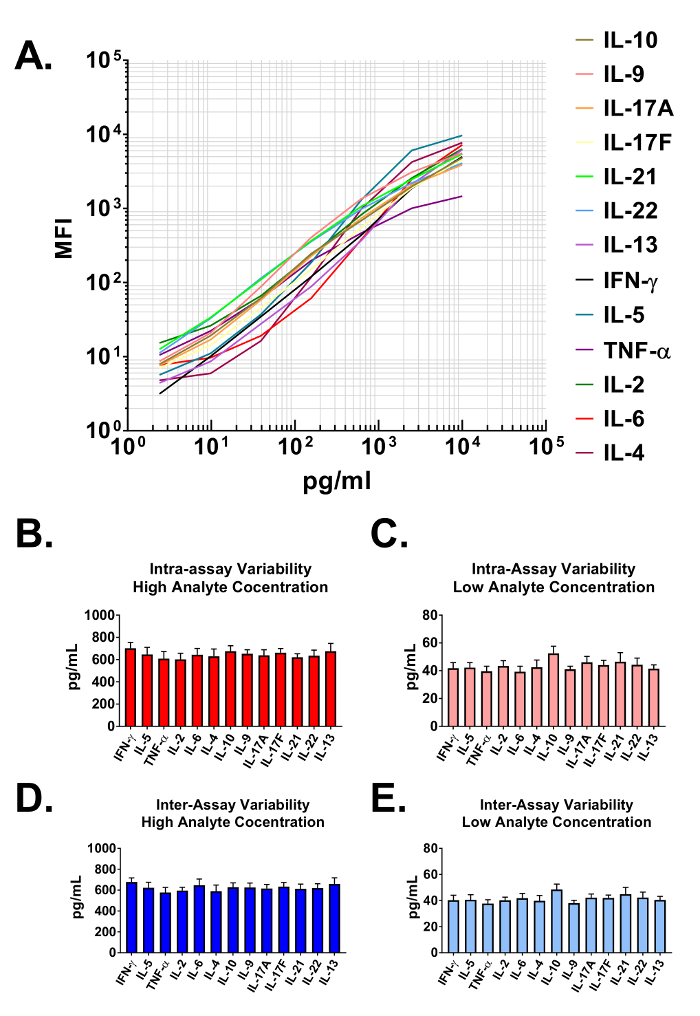
Figure 2: Standard Curve Ranges and Assay Precision. (A) A representative standard curve generated using the Mouse T helper cytokine panel. A standard curve must be run with each assay. (B-C) Intra-assay precision. Two samples with different concentrations of target proteins (high and low) were analyzed in one assay with 16 replicates for each sample. Data are represented as the mean + standard deviation. (D-E) Inter-assay precision. Two samples with different concentrations of target proteins (high and low) were analyzed in three independent assays with 3 replicates for each sample. Data are represented as the mean + standard deviation. Please click here to view a larger version of this figure.
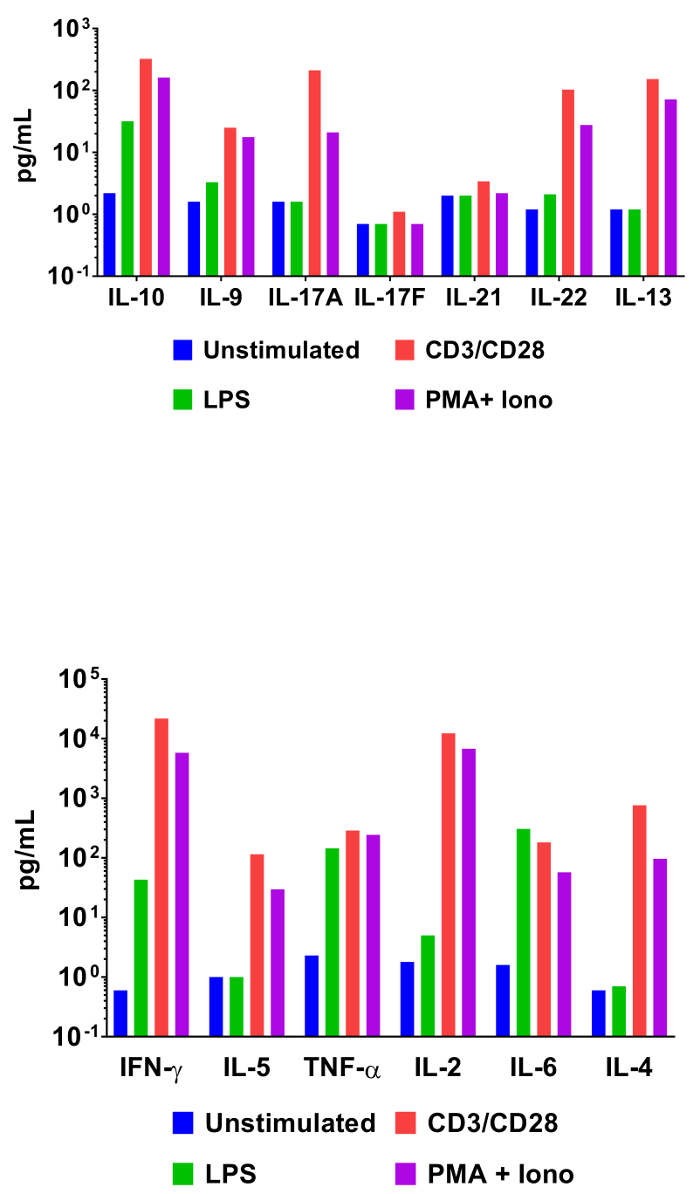
Figure 3: Quantification of Representative Results. Mouse splenocytes (1 x 106 cells) were cultured at 37 °C + 5% CO2 under various conditions: unstimulated, LPS (100 ng/mL), αCD3 (1 µg/mL plate-coated) + αCD28 (1 µg/mL soluble), PMA (20 ng/mL) + Ionomycin (500 ng/mL). Culture supernatants were collected after 48 h then quantified using the Mouse T helper cytokine panel. Differential cytokine expression profiles in response to stimulation conditions are clearly visible. Please click here to view a larger version of this figure.
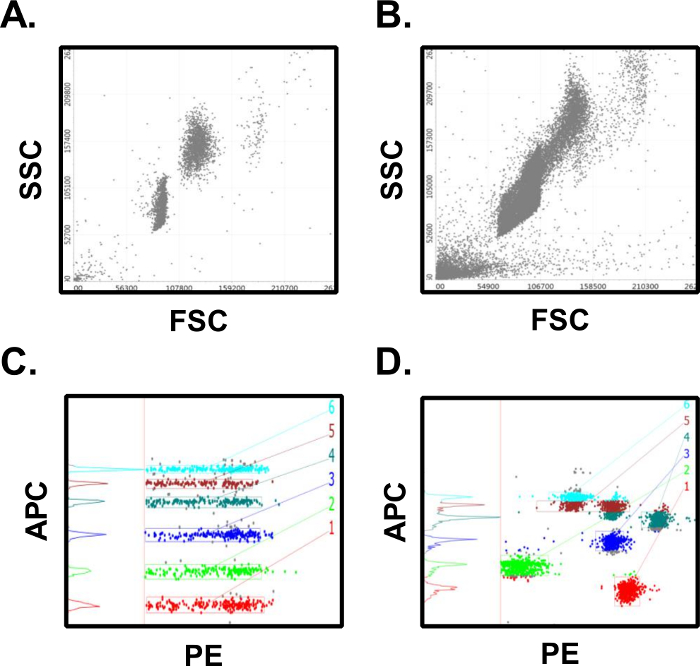
Figure 4: Results from Successful vs. Unsuccessful Assays. (A) Successful assays will have distinct bead populations for the microsphere populations. (B) Unsuccessful assays will have poor population separation, bead aggregates, and too many collected events. (C) Successful assays have clearly defined intensities in the classification fluorescence channel which are easy to gate and quantify. (D) Unsuccessful assays will have poorly separated and classification intensities which overlap multiple bead populations, making quantification difficult. Many of these errors can be avoided through careful adherence to the assay protocol. Please click here to view a larger version of this figure.
| Standard Curve | Sensitivity (pg/mL) | ||||
| Analyte | Fit Formula | CV | R2 | Min. | Max. |
| IFN-γ | 5-P.log(4.43, 14.40, 0.54, 9.11, 0.01) | 1.77% | 0.99 | 2.1 | 18105.0 |
| IL-5 | 5-P.log(4.36, 12.68, 0.64, 5.73, 0.36) | 1.32% | 1 | 1.9 | 14514.0 |
| TNF-α | 5-P.log(4.36, 11.63, 0.79, 5.45, 0.37) | 1.48% | 0.99 | 1.9 | 20109.0 |
| IL-2 | 5-P.log(4.46, 12.62, 0.78, 5.07, 0.34) | 1.34% | 1 | 1.9 | 25109.0 |
| IL-6 | 5-P.log(4.58, 11.66, 0.64, 5.77, 0.37) | 1.70% | 0.99 | 2.0 | 7022.0 |
| IL-4 | 5-P.log(4.27, 12.23, 0.62, 5.72, 0.36) | 1.36% | 1 | 2.0 | 13273.0 |
| IL-10 | 5-P.log(4.67, 11.65, 1.20, 6.28, 0.61) | 1.81% | 0.99 | 2.2 | 5190.0 |
| IL-9 | 5-P.log(4.32, 13.86, 0.86, 5.90, 0.45) | 1.82% | 1 | 1.9 | 12602.0 |
| IL-17A | 5-P.log(4.06, 14.03, 0.47, 6.52, 0.32) | 0.99% | 1 | 2.0 | 12285.0 |
| IL-17F | 5-P.log(4.52, 12.15, 0.61, 5.52, 0.34) | 0.92% | 1 | 2.0 | 14841.0 |
| IL-21 | 5-P.log(4.90, 8.03, 1.77, 4.74, 1.11) | 0.79% | 1 | 9.0 | 12943.0 |
| IL-22 | 5-P.log(4.57, 12.56, 0.63, 6.06, 0.39) | 1.16% | 1 | 2.0 | 6408.0 |
| IL-13 | 5-P.log(4.40, 11.44, 0.22, 3.19, 1.45) | 1.11% | 1 | 2.1 | 16728.0 |
Table 1: Standard Curve Fitting and Assay Sensitivity. The data analysis software was used to generate standard curves for all analytes contained in the multiplex assay. This fully automated analysis applies a robust 5-parameter curve fitting algorithm to the dilution series standards from the assay and was also used to define assay sensitivity using the theoretical limits of detection.
