In order to test the usability of ExCYT, we analyzed a curated data set published by Chevrier et al. titled 'An Immune Atlas of Clear Cell Renal Carcinoma' where the group conducted CyTOF analysis with an extensive immune panel on tumor samples taken from 73 patients11. Two separate panels, a myeloid and lymphoid panel, were used to phenotypically characterize the tumor microenvironment. The objective of our study was to recapitulate the results of their t-SNE and cluster analysis, showing that ExCYT could be used to come to the same conclusions as well as show additional methods of visualization and cluster analysis.
In the original manuscript, the group described 22 T cell clusters identified by the lymphoid panel and 17 cell clusters identified by the myeloid panel. In Figure 3 & Figure 4 of the publication, the group shows heatmaps of clusters, t-SNE plots with color-coded clustering solutions, and t-SNE heatmaps in subpanels A, B, & C. In order to perform the analysis, we obtained the manually gated data from Cytobank and sampled 2,000 events from each file or took the entire file if it had less than 2,000 events, following the analysis pipeline illustrated in the original manuscript. At this point, we sampled a total of 100,000 events via our post-gating subsampling parameter, conducted t-SNE analysis, and used a variety of clustering methods to explore the data in various ways.
First, we examined the myeloid panel by following the same analysis pipeline as the original manuscript by completing the t-SNE analysis and creating heatmaps of the various markers (Figure 3A). While the original manuscript normalized the t-SNE heatmaps to the 99th percentile of each marker, ExCYT does not do this type of normalization for its heatmaps. However, similar distributions of marker co-expression were observed as described in the original manuscript. We then applied a Network Graph-Based method of clustering the data by creating the graph with 100 k-nearest neighbors and clustering the graph via optimizing the modularity of the graph by using the Fast-Greedy implementation within ExCYT, where we found 19 sub-populations of cells (Figure 3B). When comparing the heatmap of these clusters created by ExCYT with the heatmap published in the original manuscript, we noted that we were able to identify similar clusters of myeloid cells (Figure 3C). Of note, the original manuscript identified and contrasted two sub-populations of myeloid cells that we identified in our analysis defined by HLA-DRintCD68intCD64intCD36+CD11b+ (Cluster 13) and HLA-DR+CD4+CD68+CD64+CD36–CD11b– (Cluster 18). Visualization by high-dimensional box plot of these two populations revealed statistically significant differences (Mann-Whitney) in the six markers mentioned (Figure 1D).
Next, we analyzed the lymphoid panel with a more conventional and faster hierarchical clustering approach. This approach yielded similar marker distributions via t-SNE heatmaps (Figure 4A). Furthermore, clustering of the data via hierarchical clustering (Figure 4B), demonstrated similar clusters of lymphoid cells (Figure 4C). Of note, we also identified the unique regulatory T cell population from the original manuscript defined as CD4+CD25+Foxp3+CTLA-4+CD127– (Cluster 17) via our high-dimensional flow plot (Figure 4D).
Finally, we wanted to employ a method within ExCYT to quickly and quantitatively assess co-associations among markers. We began by using a hard k-means clustering algorithm to lay down 5,000 clusters on the two-dimensional t-SNE data (Figure 4E). We then used the median expression of all the markers of all these clusters to create a heatmap from these clusters (Figure 4F). Since these heatmaps cluster rows as well as columns that are similar, this method of abstracting the data by applying a fine mesh of clusters and then creating a heatmap allows us to pick up co-associations easily, such as the co-association of Tim-3, PD-1, CD38, and 4-1BB.
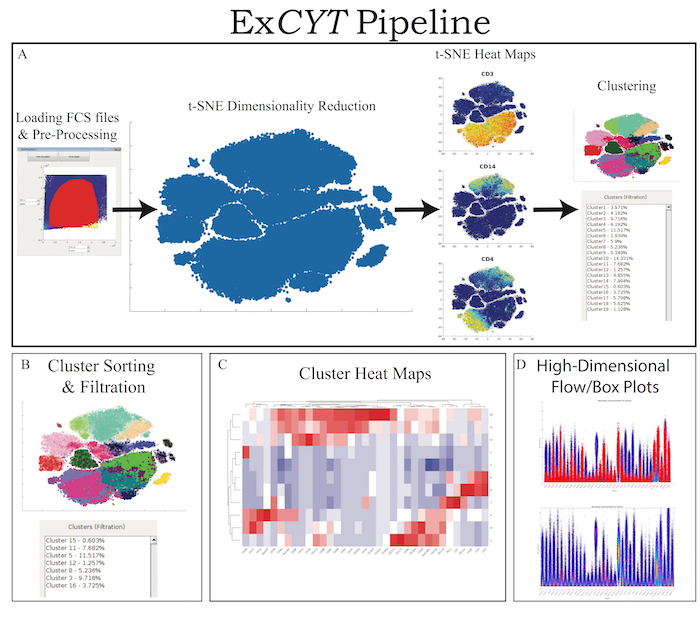
Figure 1: ExCYT Pipeline & Features. (A) ExCYT begins by importing raw FCS data, applying optional compensation, gating, and random subsampling prior to downstream analysis. This ensures all events being analyzed are relevant to the experiment being analyzed. t-SNE dimensionality reduction is then performed to visualize all events and t-SNE heatmaps can be generated to visualize phenotypic distributions. Finally, a variety of clustering algorithms can be applied on either t-SNE transformation or high-dimensional raw data. (B) Novel sorting and thresholding features allow users to quickly sort through possibly hundreds of clusters to find ones of interest. (C) Heatmaps of clusters can be created to examine how multiple clusters compare to each other as well as which markers co-associate. (D) Novel high-dimensional flow/box plots can be generated as a form of back-gating clusters on original data while appreciating the high-dimensional nature of the data. Please click here to view a larger version of this figure.
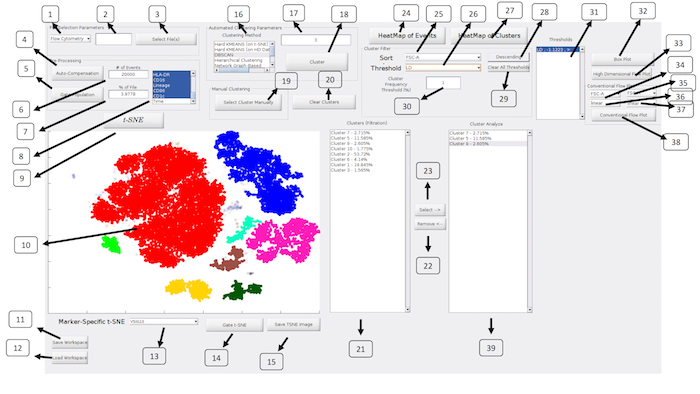
Figure 2: ExCYT Graphical User Interface: The ExCYT graphical user interface allows for a streamline work flow working from the left to right of the panel as the user imports their data, conducts t-SNE dimensionality reduction, clustering, and final cluster analysis and visualization. Please click here to view a larger version of this figure.
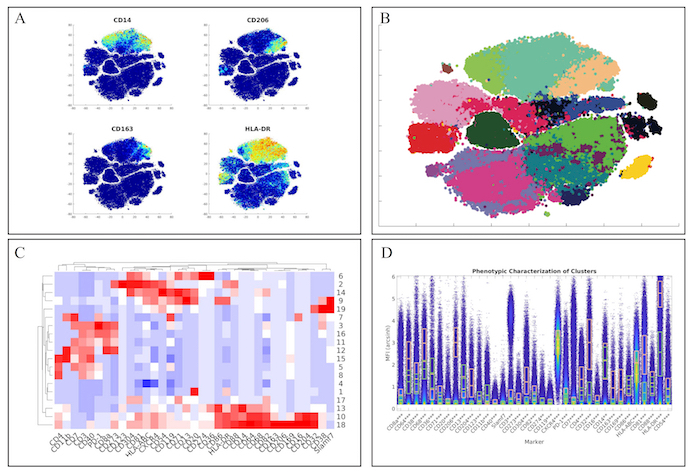
Figure 3: Recapitulation of Myeloid Sub-Populations from Chevrier et al. (A) Token t-SNE heatmaps of myeloid panel (B) t-SNE plot of myeloid panel color coded by Network-Graph clustering algorithm (C) Heatmap of clusters identified by clustering solution on myeloid panel (D) Comparative high dimensional box plot comparing contrasting myeloid subpopulations (Clusters 13 & 18) referenced in original manuscript Please click here to view a larger version of this figure.
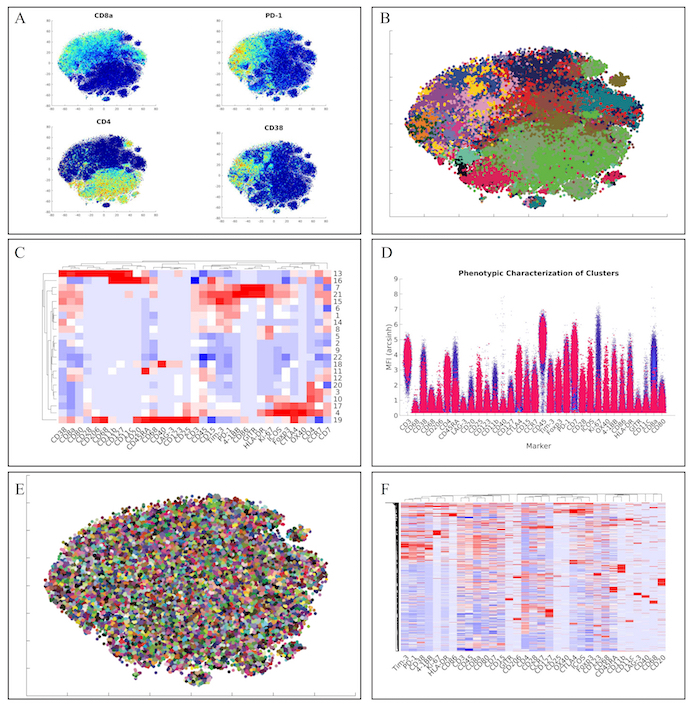
Figure 4: Recapitulation of Lymphoid Sub-Populations from Chevrier et al. (A) Token t-SNE heatmaps of lymphoid panel (B) t-SNE plot of lymphoid panel color coded by hierarchical clustering algorithm (C) Heatmap of clusters identified by clustering solution on lymphoid panel (D) High dimensional flow plot of identified regulatory T cell population (Cluster 17) in original manuscript (E) Clustering solution of 5,000 cluster hard k-means analysis on t-SNE data (F) Heatmap of clusters identified by k-means clustering solution on lymphoid panel showing marker co-associations. Please click here to view a larger version of this figure.
| No. | Description | Name (in GUI) | |
| 1 | Select type of Cytometry | NA | |
| 2 | Random subsampling of raw data | NA | |
| 3 | Select files for analysis | Select File(s) | |
| 4 | Auto-compensation of raw data based on directory of single stains provided to software | Auto-Compensation | |
| 5 | Gating to select events for t-SNE and clustering analysis | Gate Population | |
| 6 | Random subsampling of gated data (absolute number) | NA | |
| 7 | Random subsampling of gated data (percent of gated population) | NA | |
| 8 | Select channels for analysis | NA | |
| 9 | Run t-SNE dimensionality reduction | t-SNE | |
| 10 | t-SNE Window | NA | |
| 11 | Save workspace | Save Workspace | |
| 12 | Load Workspace | Load Workspace | |
| 13 | Create t-SNE heatmap on select marker | NA | |
| 14 | Gate t-SNE to re-do t-SNE analysis of select population | Gate t-SNE | |
| 15 | Save t-SNE window as image | Save TSNE Image | |
| 16 | Select Clustering Algorithm | Clustering Method | |
| 17 | Enter Clustering Parameter for given algorithm | NA | |
| 18 | Cluster Analysis | Cluster | |
| 19 | Draw Clusters Manually | Select Cluster Manually | |
| 20 | Clear All Clusters to redo cluster analysis | Clear Clusters | |
| 21 | Show Clusters under current filter conditions | Clusters (Filtration) | |
| 22 | Remove select clusters from Cluster Analyze listbox | Remove <– | |
| 23 | Add cluster to Cluster Analyze listbox | Select –> | |
| 24 | Create conventional heatmap of all events in analysis | HeatMap of Events | |
| 25 | Sort clusters by select marker | Sort | |
| 26 | Set threshold by select marker | Threshold | |
| 27 | Create conventional heatmap of select clusters from Cluster Analyze listbox | HeatMap of Clusters | |
| 28 | Flip order of sort | Ascending/Descending | |
| 29 | Clear all thresholds | Clear All Thresholds | |
| 30 | Set frequency threshold for clusters | Cluster Frequency Threshold (%) | |
| 31 | List of current thresholds active on 'Clusters (Filtration)' listbox | Thresholds | |
| 32 | High Dimensional Box Plot | High Dimensional Box Plot | |
| 33 | High Dimensional Flow Plot | High Dimensional Flow Plot | |
| 34 | Horizontal axis parameter for conventional flow plot | NA | |
| 35 | Vertical axis parameter for conventional flow plot | NA | |
| 36 | Data transformation for conventional flow plot on horizontal axis | NA | |
| 37 | Data transformation for conventional flow plot on vertical axis | NA | |
| 38 | Create conventional flow plot | Conventional Flow Plot | |
| 39 | Show Clusters for Analysis | NA | |
Table 1: Overview of All Functions Present in the ExCYT GUI
| Name of Software/Package | ExCYT | CYT | FCS Express | flowCore | openCyto | FlowMeans |
| Program Type | Matlab | Matlab | Stand-Alone Application | R | R | R |
| Price to User | Free | Free | $1,000 | Free | Free | Free |
| Graphical User Interface | Yes | Yes | Yes | No | No | No |
| Dimensionality Reduction Techniques | t-SNE | t-SNE,PCA | t-SNE, PCA, SPADE | none | none | none |
| Clustering Algorithms | K-Means DBSCAN Hierarchical Clustering Self-Organized Map Multiple Network-Graph Based Methods GMM – EM GMM – Variational Bayesian Inference |
K-Means GMM – EM Single Network-Graph Based Method (Phenograph) |
K-Means | none | automation of manual gating workflow | K-Means |
| Ability to Sort/Filter Clusters | Yes | No | No | No | No | No |
| High Dimensional Flow Plots | Yes | No | No | No | No | No |
Table 2: Overview of Software-assisted Flow Cytometry Analysis Solutions
