Naive CD4+ T cells isolated from two 8-week-old male C57BL/6 mice were divided into three portions. One portion of the cells was differentiated into THGM cells following the protocol described. Another portion was cultured under a THGM condition in the presence of anti-IL-4 antibody (10 µg/mL) to test the influence of an IL-4 blockade in the differentiation of THGM. The last portion was cultured under a TH17 differentiation condition (3 µg/mL anti-CD3e, 1 µg/mL anti-CD28, 10 ng/mL TGFβ, 30 ng/mL IL-6, 10 µg/mL anti-IFNγ, and 10 µg/mL anti-IL-4). After 3 days of differentiation, the cells were harvested and restimulated to analyze the cytokine expression by intracellular cytokine staining, qPCR, and ELISA. Results from the intracellular cytokine staining and FACS analysis demonstrated that about 55% of the cells cultured under the THGM differentiation condition were GM-CSF-expressing cells (Figure 1A), whereas only about 2% of the cells differentiated under the TH17 condition expressed GM-CSF. In addition, compared to the TH17 differentiation condition which generated 8.17% IL-17-producing cells, the THGM differentiation condition only resulted in about 1% of IL-17-expressing cells. Under the two differentiation conditions tested, only a small fraction of the cells expressed IFNγ (<1%). Furthermore, few IL-4-expressing T cells (<1%) were seen in THGM or TH17 culture (Figure 1B). These results showed that using the described THGM cell differentiation condition, we have successfully generated T helper cells that predominantly express GM-CSF.
The successful differentiation of THGM cells was further confirmed by results from qPCRs and ELISA assays to examine the expression of GM-CSF and IL-17 at both RNA and protein levels in the differentiated cells. The generated THGM cells had a significantly higher expression of Csf2 but a much lower expression of Il17 compared to the TH17 cells (Figure 2A). As shown in Figure 2B, GM-CSF protein was detected in culture supernatants from both THGM and TH17 cells. Nevertheless, the GM-CSF concentration in the THGM culture supernatant was about threefold of that in TH17 cells. In addition, proteins of IL-17 (Figure 2B), IFNγ, and IL-4 were undetectable in the THGM cell culture supernatant. Since RORγt has been identified as the master transcription factor of TH17 cells, while STAT3 and STAT5 have been shown to have a great importance in the development of TH17 and THGM cells, respectively, we examined their mRNA expression in the THGM and TH17 cells. It was found that the THGM cells had a significantly lower expression of Rorc and a higher Stat5 expression than the TH17 cells (Figure 3). Interestingly, the THGM cells had a slightly lower (but not significant) expression of the Stat3 gene compared to the TH17 cells. These results indicated that although the THGM and the TH17 differentiation are governed by different transcriptional factors, they may share some common features.
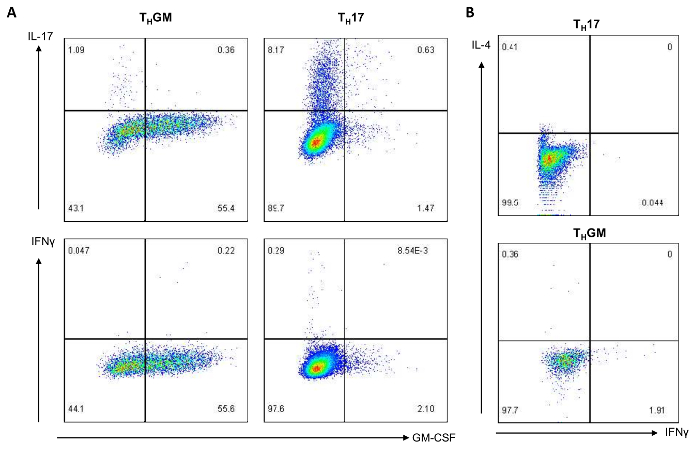
Figure 1: THGM cells predominantly express GM-CSF. THGM and TH17 cells were harvested on day 3 after the differentiation. (A) For 5 h, the cells were restimulated with PMA and ionomycin in the presence of a protein transport inhibitor. The expression of GM-CSF, IL-17, and IFNγ was analyzed by intracellular cytokine staining followed by flow cytometry. (B) The IL-4 and IFNγ expression by TH17 or THGM cells was analyzed. Please click here to view a larger version of this figure.
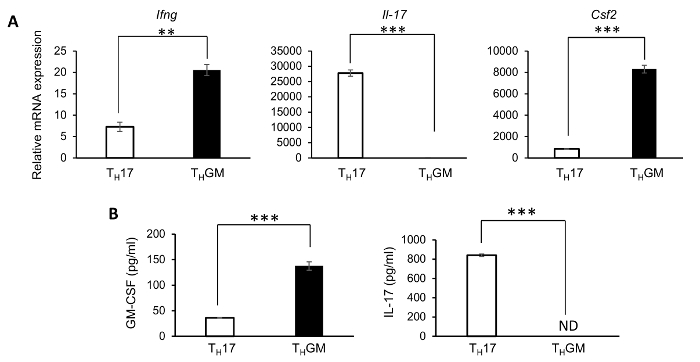
Figure 2: Expression of IL-17, IFNγ, and GM-CSF in TH17 and THGM cells. THGM and TH17 cells were harvested and restimulated with plate-bound anti-CD3e, 3 days after the differentiation. (A) Cells were harvested to isolate RNA for cDNA preparation, 3 h after the stimulation. The expressions of Ifng, Il-17, and Csf2 were determined by a quantitative real-time PCR (qPCR). (B) The culture supernatant was harvested at 24 h after the stimulation, to determine the concentration of GM-CSF in THGM cells, or IL-17 in TH17 cells, by ELISA. (** p <0.01, *** p <0.001.) Please click here to view a larger version of this figure.
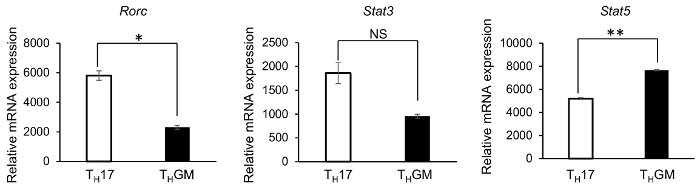
Figure 3: Rorc, Stat3, and Stat5 gene expression in TH17 and THGM cells. Differentiated THGM and TH17 cells were harvested and restimulated with plate-bound anti-CD3e for 3 h. Total RNA was isolated to prepare cDNA. The Rorc, Stat3, and Stat5 expressions were determined by qPCR. (* p <0.05, ** p <0.01; NS = not significant.) Please click here to view a larger version of this figure.
| Name | Stock concentration | working concentration | Dilution | volume in 500 μL staining buffer |
| CD4-PerCp | 0.2 mg/mL | 2.8 μg/mL | 1:71 | 7 μL |
| CD44-APC | 0.2 mg/mL | 2.4 μg/mL | 1:83 | 6 μL |
| CD25-PE | 0.2 mg/mL | 2 μg/mL | 1:100 | 5 μL |
| CD62L-FITC | 0.5 mg/mL | 10 μg/mL | 1:50 | 10 μL |
| GM-CSF-PE | 0.2 mg/mL | 2 μg/mL | 1:100 | |
| IL-17A-FITC | 0.5 mg/mL | 5 μg/mL | 1:100 | |
| IL-17A-APC | 0.2 mg/mL | 2 μg/mL | 1:100 | |
| IFNγ-APC | 0.2 mg/mL | 2 μg/mL | 1:100 | |
| IFNγ-PE | 0.2 mg/mL | 2 μg/mL | 1:100 | |
| IL-4-APC | 0.2 mg/mL | 2 μg/mL | 1:100 | |
| CD4-FITC | 0.5 mg/mL | 5 μg/mL | 1:100 | |
| IL-7 | 20 μg/mL | 2 ng/mL | 1:10000 | |
| Anti-CD28 | 0.5 mg/mL | 1 μg/mL | 1:500 | |
| Anti-IFNγ | 1 mg/mL | 10 μg/mL | 1:100 |
Table 1: Used cytokines and antibodies.
| Primer | Sequence | |
| Ifng Forward | 5’-TCAAGTGGCATAGATGTGGAAGAA-3’ | |
| Ifng Reverse | 5’-TGGCTCTGCAGGATTTTCATG-3’ | |
| Il-17 Forward | 5’-CTCCAGAAGGCCCTCAGACTAC-3’ | |
| Il-17 Reverse | 5’-AGCTTTCCCTCCGCATTGACACAG-3’ | |
| Csf2 Forward | 5’-TTTACTTTTCCTGGGCATTG-3’ | |
| Csf2 Reverse | 5’-TAGCTGGCTGTCATGTTCAA-3’ | |
| Rorc Forward | 5’-TTTGGAACTGGCTTTCCATC-3’ | |
| Rorc Reverse | 5’-AAGATCTGCAGCTTTTCCACA-3’ | |
| Stat3 Forward | 5’-TGGCCCTTTGGAATGAAGGGTACA-3’ | |
| Stat3 Reverse | 5’-CACTGATGTCCTTTTCCACCCAAGT-3’ | |
| Stat5 Forward | 5’-TGCCCGGCTGGAACTACACCTT-3’ | |
| Stat5 Reverse | 5’-ATGCCCCCGATTTCCGAGTCAC-3’ | |
Table 2: Primers for qPCR.
| step | Temp. (°C) | time | repeat | |
| 1 | 95 | 2 min | ||
| 2 | 95 | 3 s | ||
| 3 | 60 | 30 s | step 2 and 3, 39 times | read plate |
| 4 | 65–95 | 5 s | step 4, 30 times, +0.5°C each repeat | read plate |
Table 3: PCR program to assess gene expression.
