Human induced pluripotent stem cells were cultured, and the DNA damage and the tail moment, which were used as a measure of DNA integrity, were analyzed by comet assay. iPS cells were embedded in low-melting-point agarose and placed on a glass slide. The cells were, then, treated with lysis buffer, followed by an alkaline solution, to obtain supercoiled DNA. Nucleoids were electrophoresed and comets were visualized by DNA dye (Figure 1A-D). The comets were, then, analyzed with ImageJ, using the comet assay plugin (Figure 2A-C).
Human iPS cells were treated with Doxo to induce DNA damage and to be used as a positive control. Representative micrographs of comets from Doxo-treated and nontreated iPS cells are shown in Figure 3A. A basal amount of DNA damage was found in iPS cells, expressed as a fraction of DNA damage and tail moment. However, the Doxo treatment increased the DNA damage in iPS cells as expected (Figure 3B,C). This shows that the comet assay can be used to assess DNA integrity not only in somatic cells18,19 but also in pluripotent stem cells21.
Freshly differentiated iPS cardiomyocytes (iPS-CMs), iPS-CMs cultured for 6 months (prolonged culture [PC]), and iPS-CMs treated with Doxo were subjected to γH2A.X immunolabeling. Representative micrographs of γH2A.X immunolabelling are shown in Figure 4A (lower-magnification) and Figure 4B (higher-magnification). The number of γH2A.X foci (DDR foci) (punctae) in each nucleus are quantified, using CellProfiler with custom pipeline modules (Figure 5A-L). The percentage of cells that are positive for γH2A.X are classified into nuclei with zero to five punctae and nuclei with more than 10 punctae. In the control iPS-CM, more than 90% of the cells had less than five DDR foci per nuclei, and a total of less than 10% of the cells had more than six DDR foci per nuclei (Figure 4C, control [Ctrl]). iPS-CMs cultured for 6 months had less than 90% cells with less than five DDR foci per nuclei, and a total of more than 13% of the cells had more than six DDR foci per nuclei (Figure 4C, PC), whereas the Doxo-treated iPS-CM showed less than 80% of the cells with less than five DDR foci per nuclei, and a total of about 24% of the cells had more than six DDR foci per nuclei (Figure 4C, Doxo). This data clearly shows that prolonged cell culture and Doxo treatment induce significant DNA damage in iPS-CMs and are not suitable for cell transplantation.
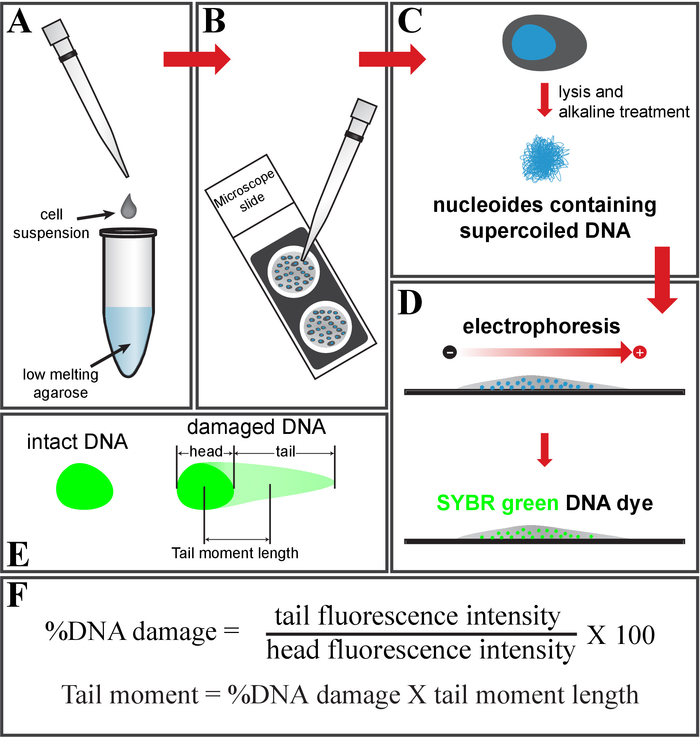
Figure 1: Schematic of the comet assay. (A) Mix the cell suspension with low-melting-point agarose and (B) place it on a glass slide. (C) Treat it with cell lysis buffer, followed by an alkaline solution, to get nucleoids containing supercoiled DNA. (D) Electrophorese and stain the DNA using SYBR green DNA dye. (E) Schematic of intact (left) and damaged DNA (right, comet shape). (F) Formula to calculate a fraction of the DNA damage and tail moment. Please click here to view a larger version of this figure.
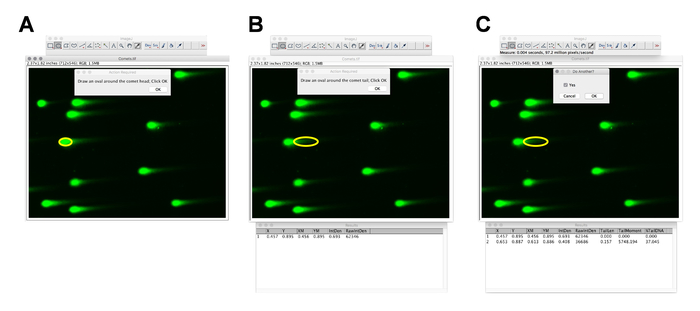
Figure 2: DNA damage and tail moment quantification by ImageJ. (A–C) Screenshots of ImageJ, with the comet analysis plugin showing a selection of the comet head and tail. Please click here to view a larger version of this figure.
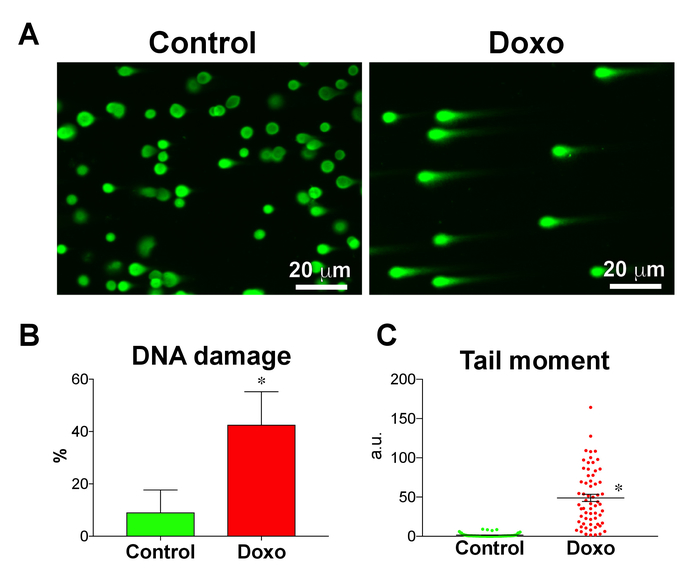
Figure 3: Doxorubicin induces DNA damage in human iPS cells. (A) DNA damage in doxorubicin-treated (Doxo) and nontreated (control) iPS cells, analyzed using the comet assay. (B) Fraction of DNA damage (n = 3) and (C) tail moment (n = 63 comets) quantified using the comet assay. Treatment with doxorubicin, a DNA-intercalating agent, significantly increased the fraction of DNA damage and tail moment in iPS cells. Data are means ± SEM. *P < 0.05, Student's unpaired t-test. Please click here to view a larger version of this figure.
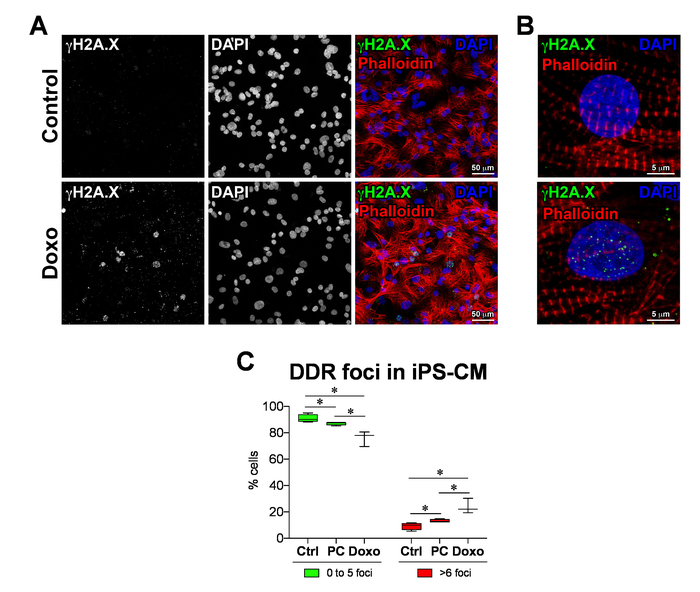
Figure 4: DNA damage response in iPS-derived cardiomyocytes. (A) DNA damage response marker γH2A.X identified by immunofluorescence in doxorubicin-treated (Doxo) and nontreated (control) iPS-cardiomyocytes as well as prolonged culture iPS-cardiomyocytes (PC). (B) Higher magnification of γH2A.X (green punctae) labeling at the sites of DNA damage in iPS-CMs. (C) Quantification of DDR foci, analyzed using CellProfiler with custom pipeline modules. Data are means ± SD; n = 3 to 4. *P < 0.05, Student's unpaired t-test. Please click here to view a larger version of this figure.

Figure 5: Automated DNA damage response analysis by CellProfiler. (A–L) Screenshots of CellProfiler with specified settings for importing the images, and the automated identification and quantification of γH2A.X punctae in iPS-CMs. Please click here to view a larger version of this figure.
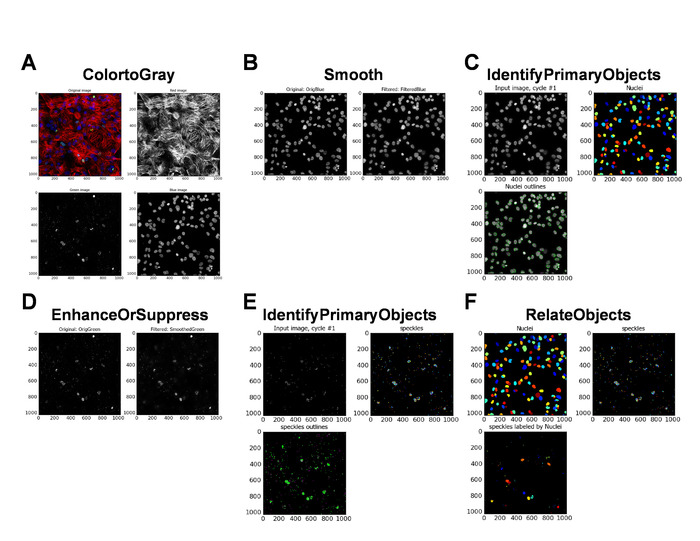
Figure 6: Automated DNA damage response analysis by CellProfiler. (A–F) Data images generated by CellProfiler at the completion of the image analysis. Please click here to view a larger version of this figure.