The sizing results of the premutation female reference sample (NA20240, repeat sizes of 30 and 80) and the full mutation female reference sample (NA20239, repeat sizes of 20 and 200) are shown in Figure 1A and Figure 1B, respectively. Typically, two marker peaks (lower marker 50 base pairs [bp] and upper marker 10,380 bp) are included in the fragment size profile. There is usually a primer complex peak with a size of nearly 95 bp. Through the reference sample, a linear regression standard curve with four points can be constructed, as exhibited in Figure 1C.
The representative size features of clinical normal, intermediate, premutation, and full mutation samples are shown in Figure 2A–D. Particularly, mosaic full mutations with two expanded fragment peaks and one normal peak are presented in Figure 2E. In some female cases, only one peak is displayed in the microfluidic electrophoresis results, as shown in Figure 2F. This can be explained by the presentation of normal homozygous alleles (with the same CGG repeat numbers in the two alleles) in these cases or the inability of differentiating heterozygous alleles that have repeat number differences of four or less29. However, these single-peak results could be classified as normal, because it has been validated that the PCR-based pipeline enables robust amplification and detection of full mutation or premutation alleles, which minimizes the possibility of false negative in this situation. One type of sub-optimal situation is baseline bias, as shown in Figure 2G, which could produce ambiguous or uninterpretable results in some cases. Such a condition is suspected to be caused by an instrument issue. The measured fragment sizes of unknown samples are plotted against the linear regression standard curve derived from the reference samples to automatically calculate the repeat sizes using the analysis software, as displayed in Figure 2H. Fragment sizes less than 200 repeats are interpolated into the linear regression standard curve while larger full-mutation allele sizes are measured by extrapolating along the same standard line. The software also displays the classification of each sample according to different guidelines.
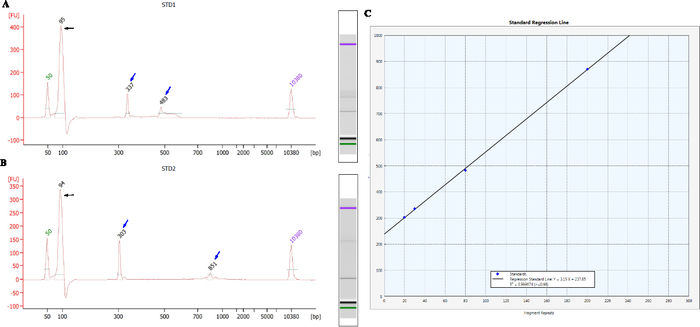
Figure 1: The sizing results of the female reference samples and the corresponding linear regression curve. (A, B) show the size features of the premutation female reference sample (NA20240, repeat sizes of 30 and 80) and the full mutation female reference sample (NA20239, repeat sizes of 20 and 200), respectively. Two markers (lower marker, 50 bp and upper marker 10380 bp) are included in the electrophoresis result for each sample. The peak with a size of nearly 95 bp indicates a primer complex. Black arrows indicate the peak size of primer complex, and blue arrows represent the fragment length of reference sample. (C) The linear regression standard curve with four points (blue diamonds) from the two reference samples is constructed in the analysis software. The horizontal and vertical axes show the calculated CGG repeat numbers and the measured fragment length in microfluidic electrophoresis. The R2 value of the regression was 0.99967. Please click here to view a larger version of this figure.
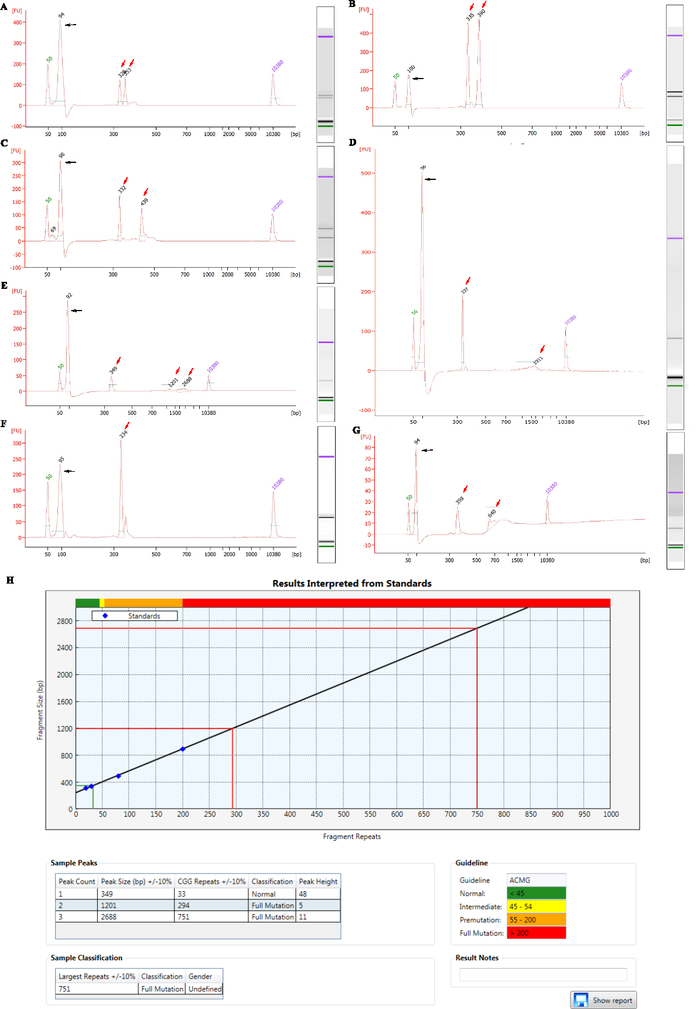
Figure 2: The representative results of clinical samples with different CGG repeat size. (A–E) show the representative size features by bioanalyzer in the order of normal (with fragment lengths of 328 bp and 353 bp, corresponding to the repeat numbers of 28 and 36), intermediate (with fragment lengths of 335 bp and 390 bp, corresponding to the repeat numbers of 30 and 49), premutation (with fragment lengths of 332 bp and 439 bp, corresponding to the repeat numbers of 29 and 65), full mutation (with fragment lengths of 337 bp and 1911bp, corresponding to the repeat numbers of 31 and 545) and mosaic full mutations (with fragment lengths of 349 bp, 1201 bp and 2688 bp, corresponding to the repeat numbers of 33, 294 and 751) samples. (F) presents a single-peak microfluidic electrophoresis result (with a fragment length of 334 bp, corresponding to the repeat number of 30) of a female who probably has homozygous alleles (with the same CGG repeat numbers in the two alleles) or heterozygous alleles (with the CGG repeat number differences of four or less). (G) shows one type of the sub-optimal situation that is baseline bias. Black arrows indicate the peak size of primer complex, and red arrows represent the fragment length of the sample. (H) displays the main result interface of the analysis software. The measured fragment sizes of unknown samples are plotted against the linear regression standard curve to automatically calculate the repeat sizes. The normal allele of the mosaic full-mutation female sample shown in (E) is mapped to the standard curve at the lower left corner of the coordinate axis region (plotted in green), and the larger full-mutation alleles (plotted in red) extrapolated beyond the four standard points (blue diamonds on the curve). The tabular sections in the lower half of the figure present the fragment sizes and the corresponding diagnostic classification according to the chosen ACMG guideline boundaries. Please click here to view a larger version of this figure.
