Representative results from both the monocyte subset panel and T-cell subset panel are described below.
Figure 1 illustrates the hierarchical gating strategy for the monocyte subset panel on immune cells gathered from bone marrow of DMM treated animals. The same strategy was used and verified in all other tissue types. When setting up the experiment, the Forward Scatter Area (FSC-A) and Side Scatter Area (SSC-A) voltage was determined for each tissue type to identify monocytes/macrophages and exclude T-cells and debris (G1). During each experiment, unstained controls of each tissue type were analyzed, and FSC-A and SSC-A voltage adjusted when necessary. Voltages are expected to stay similar over time, if parameters change drastically a blockage of the cytometer is likely. Furthermore, unstained controls were used to determine the true negatives for the dead/alive stain and gates were adjusted each time the experiment was conducted accordingly (Figure 2A). When designing the experiment, fluorochromes should be chosen carefully, and normally surface markers with low expression are paired with bright fluorochromes (e.g., here Alexa Fluor 647 was used for CD206). Various dead/alive stains exist that can be detected by different wavelengths; here, FVS510 was used.
Figure 3 illustrates sample data from immune cells isolated from synovial tissues and stained with extracellular surface markers 6 weeks after animals received either DMM or sham-control surgery. All subsets can easily be identified using the protocol both in study and control animals. In particular, differences between groups can be seen for macrophage subsets (higher percentage of Ly-6C+/MHC-II- macrophages (G7) in the DMM group) and the expression of M1 and M2 macrophages (higher percentage of M2 macrophages in the DMM group).
Figure 4 visualizes the hierarchical gating strategy for the extra- and intracellular T-cell panel on immune cells isolated from the spleen of DMM treated animals. Principles are identical to the ones used for the monocyte panel. However, the fixing and permeabilization process changes the size and density of cells. Thus, typical FSC and SSC parameters need to be determined using a back-gating process from CD3+ cells when first setting up the experiment for each cell type. Some fluorochromes tend to aggregate over time (e.g., PE that was used with FoxP3 here). Aggregates can potentially modify the results due to the high brightness that influences the spectral-overlap and compensation. Thus, all antibodies were vortexed and spun down each time prior to their use in order to decrease aggregates. Additionally, a gating strategy was used to further reduce the influence of aggregates (G2). While setting up the experiments fluorescence minus one controls (FMOs) were performed for each antibody. Sample data is shown in Figure 2B,2C.
Figure 5 and Figure 6 show immune cells that were isolated from lymph nodes (Figure 5) and synovial tissue (Figure 6) and stained using the T-cell panel protocol 4 weeks after animals received either DMM or sham-control surgery. The data shows a higher percentage of Th1 cells in DMM animals (G9) in both tissues. Furthermore, intracellular staining for T-regulatory cells (G11) and Th17 cells (G12) is successful using the protocol and differences can be detected between groups.
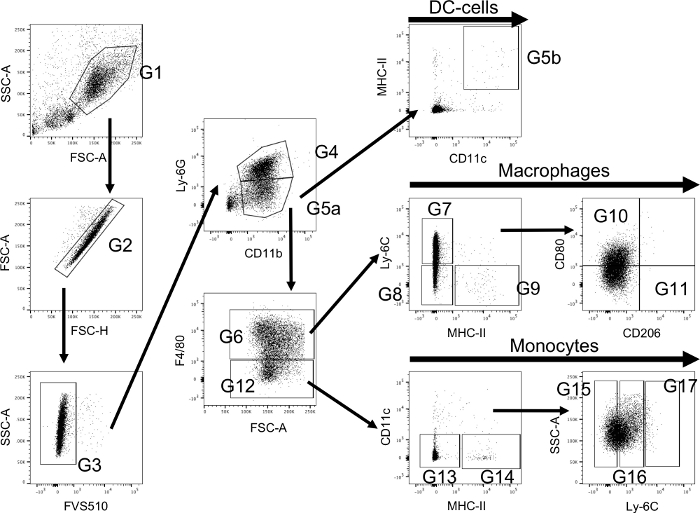
Figure 1: Flow cytometry hierarchical gating strategy using extracellular staining to identify monocytes/macrophages and their subsets. Myeloid cells are primarily identified using a forward/side scatter (FSC-A and SSC-A) dot plot (G1). Thereafter, singlets are detected using FSC-A and FSC-H (G2) and afterwards live cells are selected (G3). Cells from G3 are further classified using Ly-6G to identify neutrophils (G4) and CD11b for monocyte/macrophages (G5a). MHC-II is used to identify dendritic cells (G5b) amongst CD11b positive cells and F4/80 is used to select between macrophages (G6) and monocytes (G12). Macrophages are further classified into their subsets using Ly-6C and MHC-II (Ly-6C+/MHC-II- macrophages (G7); Ly-6C-/MHC-II- tissue resident macrophages (G8); Ly-6C-/MHC-II+ blood originated macrophages (G9)). More subsets can be selected from the entirety of macrophages and its respective subsets using CD206 and CD80 (M1: CD80+/CD206- (G10); M2: CD80-/CD206+ (G11)). Monocytes are further classified using MHC-II and CD11c (MHC-II-/CD11c- monocytes (G13); MHC-II+/CD11c- monocytes (G14)). The level of activation is then classified using the expression of Ly-6C and divided into low (G15), medium (G16) and high (G17). Please click here to view a larger version of this figure.
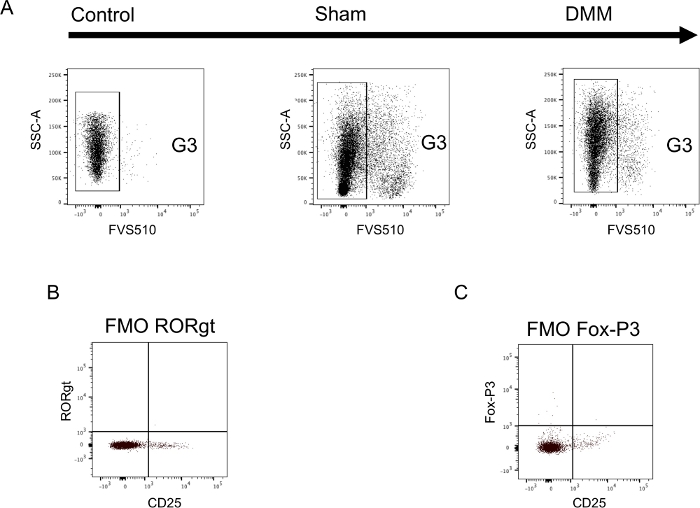
Figure 2: Sample data illustrating appropriate controls in both the monocyte and T-cell panel. (A) Synovial tissue was harvested 6 weeks after either DMM-surgery (DMM) or sham-control-surgery (sham) and a single cell suspension was stained using extracellular surface markers. During each experiment unstained cells were used to determine true negatives for the dead/alive stain and to set gates (Control). Setting of gates using unstained cells is shown in panel A. (B+C) Spleen cells were harvested from untreated control animals, a single cell suspension generated and stained using both extra- and intracellular markers. Fluorescents-minus-one (FMO) controls were generated by staining cells with the entire antibody panel missing only one antibody. Sample data is shown for both intracellular antibodies. (B) FMO -RORgt and (C) FMO -Fox-P3. FMOs were performed for both panels and used to set each gate. Please click here to view a larger version of this figure.
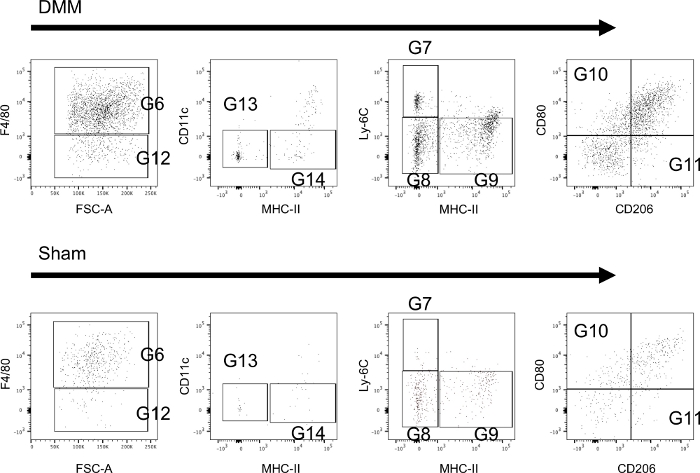
Figure 3: Extracellular staining of monocytes/macrophages isolated from the synovium of mice. Sample tissues were collected 6 weeks after mice received either DMM-surgery (DMM) or sham-control-surgery (sham). Further information regarding the utilized gates can be found in Figure 1. Sample data shows that all subsets can be reliably identified and differences can be seen between groups. Please click here to view a larger version of this figure.
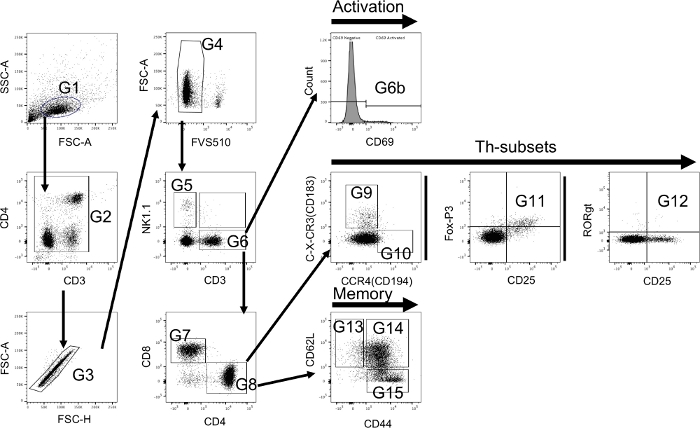
Figure 4: Flow cytometry hierarchical gating strategy using both extra- and intracellular staining to identify T-cells and their subsets. T-cells are primarily identified using a forward/side scatter (FSC-A and SSC-A) dot plot (G1). Due to the nature of the utilized antibodies aggregates should be excluded using CD3 and CD4 (G2). Thereafter, singlets are detected using FSC-A and FSC-H (G3) and afterwards live cells are selected (G4). Cells from G4 are further classified using NK1.1 to identify natural killer cells (G5) and CD3 to identify T-cells (G6). The level of activation is determined using CD69 (G6b). Thereafter, CD4 and CD8 are used to identify T-killer cells (CD4-/CD8+ (G7)) and T-helper cells (CD4+/CD8- (G8)). T-helper cells are classified into Th-1 (C-X-CR3+/CCR4- (G9)) and Th-2 cells (C-X-CR3-/CCR4+ (G10)) using C-X-CR3 (CD183) and CCR4 (CD194). In addition, Th-17 cells (CD25+/RORgt+ (G11)) and T-regulatory cells (CD25+/Fox-P3+ (G12)) are identified using intracellular markers. Furthermore, memory cell subsets are identified from T-helper cells using CD44 and CD62L (CD44-/CD62L+ naïve T-memory cells (G13); CD44+/CD62L+ central T-memory cells (G14); CD44+/CD62L- effector T-memory cells (G15)). Please click here to view a larger version of this figure.
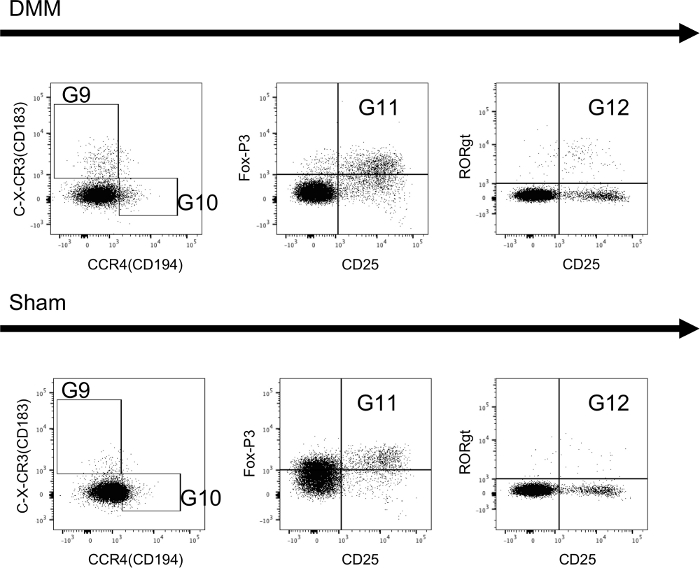
Figure 5: Extracellular and intracellular staining of T-cells isolated from draining lymph nodes of mice. Sample tissues were collected 4 weeks after mice received either DMM-surgery (DMM) or sham-control-surgery (sham). Further information regarding the utilized gates can be found in Figure 4. Sample data shows that all subsets can be reliably identified and differences can be seen between groups. Please click here to view a larger version of this figure.
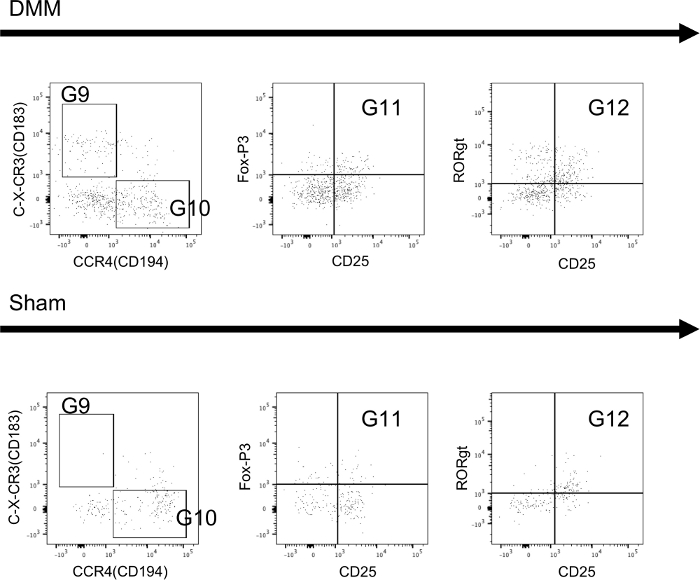
Figure 6: Extracellular and intracellular staining of T-cells isolated from synovial tissue of mice. Sample tissues were collected 4 weeks after mice received either DMM-surgery (DMM) or sham-control-surgery (sham). Further information regarding the utilized gates can be found in Figure 4. Sample data shows that all subsets can be reliably identified and differences can be seen between groups. Please click here to view a larger version of this figure.
