To obtain the most reliable data in this approach all the possible combinations should be tested (see Figure 1). In parallel, positive and negative controls should be included. The positive control should consist of the two proteins that are known to interact, of which one is fused with the larger fragment and the other is fused with the smaller fragment. The negative control ideally should consist of the two non-interacting type III membrane proteins tagged likewise. However, establishing such a control may be challenging, as the lack of interaction of the two control proteins should be thoroughly confirmed using several alternative approaches. Therefore, one might employ a cytosolic protein of non-human origin that is not expected to interact with any human protein (e.g., HaloTag) as a negative control, if N- and/or C-termini of the proteins, whose interaction is to be determined, are cytoplasmically exposed as in the case presented here. We highly recommend an additional verification of the results obtained using this type of control by co-expression of the untagged variants of either of the proteins of interest combined with titration of the corresponding plasmids. If the two proteins specifically interact, an extra copy of either of them lacking the luciferase fragment should result in a specific decrease of luminescence.
Relative luminescence units (RLU) values typical of positive and negative combinations are listed in Table 1. The results were obtained for the two NSTs, SLC35A2 and SLC35A3, that were shown to associate by co-immunoprecipitation and FLIM-FRET6 as well as in situ proximity ligation assay11. Both SLC35A2 and SLC35A3 are Golgi-resident type III membrane proteins with N- and C-termini facing the cytoplasm (Figure 1A). Therefore, there are eight possible tagging options and the resulting fusion proteins can be set together also in eight possible ways (Figure 1B). Mean RLU values corresponding to all the tested and control combinations are depicted in Figure 2A. The positive control is also included. An initial requirement for a selected result to be considered indicative of an interaction is that the RLU value obtained for the combination of interest is statistically significantly higher than the RLU value obtained for the corresponding control combination. In Figure 2A three such combinations are seen (LgBiT-SLC35A2 + SLC35A3-SmBiT, SmBiT-SLC35A2 + LgBiT-SLC35A3 and SLC35A2-SmBiT + LgBiT-SLC35A3). The next step in the analysis of results involves obtaining ratio values for the combinations of interest by dividing the corresponding RLU values by RLU values obtained for the respective controls. The results of such analysis are shown in Figure 2B. According to the manufacturer’s suggestions, ratios between 10 and 1000 are highly indicative of specific interactions. There are two combinations that meet these criteria (SmBiT-SLC35A2 + LgBiT-SLC35A3 and SLC35A2-SmBiT + LgBiT-SLC35A3) and support the idea that SLC35A2 and SLC35A3 interact. However, the fact that the other six combinations are negative does not mean that there are no interactions at all between the respective fusion proteins, but rather suggests that the tagging strategy was not optimal in these cases. This example shows how important it is to test all the possible combinations.
Data presented in Figure 2 were additionally confirmed by the co-expression of HA-tagged SLC35A2 or SLC35A3 with the combination that resulted in the highest relative luminescence, namely SLC35A2-LgBiT + SmBiT-SLC35A3. Varying amounts of the plasmids encoding HA-tagged NST variants were used for co-transfection. This resulted in a statistically significant, dose-dependent decrease in RLU values (Figure 3). Specific and dose-dependent disruption of the interaction between SLC35A2-LgBiT and SmBiT-SLC35A3 by the simultaneous co-expression of HA-tagged NST variants is shown in Table 2.
The most common problem associated with the method presented here is the poor efficiency of transfection. To verify whether this is the cause of suboptimal results, include positive control in all experiments. The positive control we employed to obtain the data presented here consists of the two interacting fusion proteins: cAPM-dependent protein kinase catalytic subunit alpha (PRKACA) fused with the smaller fragment and cAPM-dependent protein kinase type II-alpha regulatory subunit (PRKAR2A) fused with the larger fragment. In our hands, the corresponding RLU value reached ~6 x 105. A significant (2-3 orders of magnitude) drop in this number might be indicative of the suboptimal efficiency of the performed transfection. In such a case we recommend checking on the well-being of the cultured cells and making sure that the appropriate number of cells is being plated for transfection.
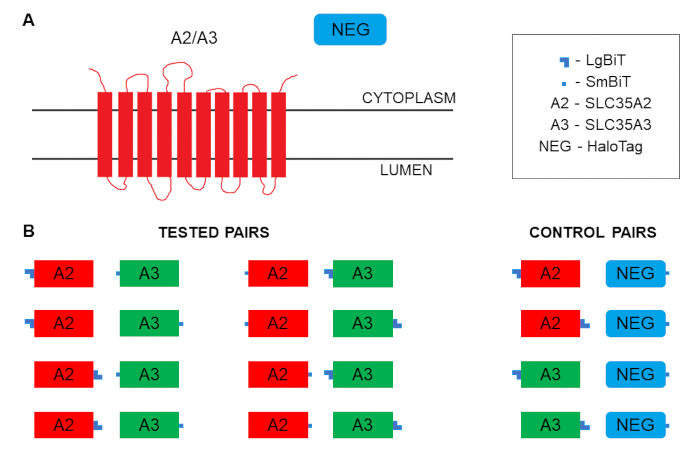
Figure 1: Schematics of membrane topology and tagging. (A) Membrane topology of SLC35A2 and SLC35A3 proteins. (B) Possibilities of tagging SLC35A2 and SLC35A3 proteins with the split luciferase complementation assay fragments and combining the resulting fusion proteins for the assay. Please click here to view a larger version of this figure.
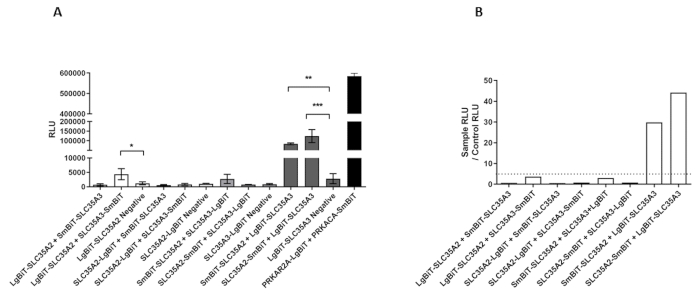
Figure 2: Results of the split luciferase complementation assay performed in HEK293T cells for the selected protein combinations and the corresponding negative controls (processed data). (A) RLU values obtained for the selected protein combinations and the corresponding negative controls. Negative, HaloTag tagged with SmBiT; PRKAR2A, cAPM-dependent protein kinase type II-alpha regulatory subunit; PRKACA, cAPM-dependent protein kinase catalytic subunit alpha. Data were analyzed using one-way ANOVA with multiple comparisons and are presented as a mean ± standard deviation (SD) from three technical replicates. p < 0.1 *, p < 0.05 **, p < 0.01 ***. (B) Fold changes calculated by dividing a mean luminescence obtained for the tested combination (RLU SAMPLE) by a mean luminescence obtained for the corresponding negative control (RLU CONTROL). Threshold value considered indicative of an interaction was set at 10. Please click here to view a larger version of this figure.
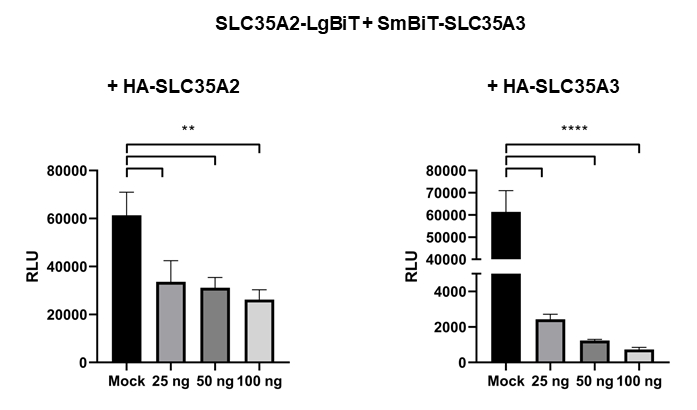
Figure 3: Specific and dose-dependent disruption of the interaction between SLC35A2-LgBiT and SmBiT-SLC35A3 by simultaneous co-expression of HA-tagged NST variants. HEK293T cells were transfected with plasmids encoding SLC35A2-LgBiT and SmBiT-SLC35A3 and, additionally, either with an empty pSelect plasmid (mock) or with increasing amounts of pSelect plasmids encoding HA-tagged SLC35A2 (left panel) and SLC35A3 (right panel). Data were analyzed using one-way ANOVA with multiple comparisons and are presented as a mean ± standard deviation (SD) from three technical replicates. p < 0.05 **, p < 0.001 ****. Please click here to view a larger version of this figure.
Table 1: Results of the assay performed in HEK293T cells for the selected protein combinations and the corresponding negative controls (raw data). Unprocessed RLU values obtained for the selected protein combinations and the corresponding negative controls are shown. Negative, HaloTag tagged with SmBiT; PRKAR2A, cAPM-dependent protein kinase type II-alpha regulatory subunit; PRKACA, cAPM-dependent protein kinase catalytic subunit alpha, TR, technical replicate. Please click here to download this table.
Table 2: Specific and dose-dependent disruption of the interaction between SLC35A2-LgBiT and SmBiT-SLC35A3 by simultaneous co-expression of HA-tagged NST variants (raw data). Unprocessed RLU values obtained for the selected protein combinations are shown. Mock, cells expressing SLC35A2-LgBiT and SmBiT-SLC35A3 co-transfected with an empty pSelect vector; HA-SLC35A2, cells expressing SLC35A2-LgBiT and SmBiT-SLC35A3 co-transfected with the indicated amounts of a pSelect plasmid encoding SLC35A2 tagged with the HA epitope at the N-terminus; HA-SLC35A3, cells expressing SLC35A2-LgBiT and SmBiT-SLC35A3 co-transfected with the indicated amounts of a pSelect plasmid encoding SLC35A3 tagged with the HA epitope at the N-terminus; TR, technical replicate. Please click here to download this table.
