Neuroblastoma is a pediatric cancer of the sympathetic nervous system arising during embryonic development or early post-natal life due to the transformation of neural crest cells1. It is the most common solid extracranial tumor in children, representing 8% of the malignancies diagnosed in patients under 15 years and is responsible for 15% of all childhood cancer deaths. The disease displays highly heterogeneous clinical behaviors due to specific chromosomal, genetic and epigenetic alterations, and histopathology features.
These alterations contribute to the aggressiveness of neuroblastoma and poor outcomes in pediatric patients. Hence, current therapies prove ineffective in the long term for almost 80% of patients with the clinically aggressive disease2, highlighting the fact that treatment for this group of patients remains challenging. This is likely due to the mechanisms of neuroblastoma heterogeneity and metastases still not being fully understood. However, the tumor microenvironment (TME) is now widely believed to play a role in the progression of many cancers; yet it remains understudied in neuroblastoma3,4.
The native TME is a complex 3D microenvironment involving cancerous and non-cancerous cells surrounded by an ECM. The ECM refers to the acellular component of a tissue that provides structural and biochemical support to its cellular residents and contributes to disease progression, patient prognosis, and therapeutic response5. This promotion of disease progression is due to "dynamic reciprocity" or ongoing bidirectional communication between cells and the ECM6,7,8. As cancer progresses, stromal collagen is reorganized often in linear patterns perpendicular to the stroma-cancer interface, which cancer cells use as a migratory route to metastasis9,10,11.
The main components of this native functional biological scaffold include a fibrous network of collagens type I and II and other proteins, including elastin, glycoproteins such as laminin, as well as a range of proteoglycans and other soluble components12,13. These proteins of the native ECM have now become attractive natural biomolecules for developing 3D in vitro models3. The application of 3D scaffolds for in vitro cell culture is increasing in popularity owing to its greater physiological representation of the TME compared to traditional 2D monolayer culture. The manufactured 3D scaffolds assist cell attachment, proliferation, migration, metabolism, and response to stimuli seen in in vivo biological systems.
The principal component of these 3D scaffolds is collagen, which is a key player in many normal biological processes including tissue repair, angiogenesis, tissue morphogenesis, cell adhesion, and migration11. Collagen-based 3D matrices have shown their robust functionality to model ECM, serving as an in vitro biomimetic microenvironment while enabling cell-ECM interactions as well as cell migration and invasion. These 3D matrices also provide a more accurate analysis of cell response to chemotherapeutic drugs than traditional 2D or "flat" culture in many cancer models14,15,16, including neuroblastoma17,18. Genetic analysis of 3D cell cultures has reported a higher correlation with the human tissue profile even when compared to animal models19. Overall, the cornerstone of these 3D scaffolds is to provide cells a suitable in vitro environment, which recapitulates the native tissue architecture and facilitates bidirectional molecular crosstalk8.
To increase the complexity of collagen-based models, other common ECM components are incorporated in the tissue engineering process, thus creating more physiologically relevant models to reflect niche TMEs of different tissues. For example, GAGs, negatively charged polysaccharides present in all mammalian tissues20, facilitate cell attachment, migration, proliferation, and differentiation. Chondroitin sulfate is a specific type of GAG found in the bone and cartilage, which has been previously used in tissue engineering applications for bone repair21,22,23,24,25. Nano-hydroxyapatite (nHA) is the main inorganic constituent of the mineral composition of human bone tissues, constituting up to 65% of bone by weight26 and is therefore widely used for bone replacement and regeneration27. Thus, GAGs and nHA are attractive composites for reconstructing the primary neuroblastoma ECM and modeling the most common metastatic sites of neuroblastoma, bone marrow (70.5%), and bone (55.7%)28.
Scaffolds incorporating these ECM components were originally developed for bone tissue engineering applications with extensive analysis of their biocompatibility, toxicity, and osteoconductive and osteoinductive features29,30. They are porous, collagen-based matrices produced using freeze-drying techniques to control their physical and biological properties. The collagen scaffolds supplemented with either nHA (Coll-I-nHA) or chondroitin-6-sulfate (Coll-I-GAG) demonstrated success in mimicking the primary TME in breast cancer31 and metastasis to bone in prostate cancer15 as well as neuroblastoma17. The freeze-drying technique used to manufacture these composite scaffolds yields reproducible homogeneity in pore size and porosity within the scaffolds22,23,24. Briefly, a collagen slurry (0.5 wt%) is fabricated by blending fibrillar collagen with 0.05 M acetic acid. For Coll-I-GAG, 0.05 wt% of chrondoitin-6-sulfate isolated from shark cartilage is added to the collagen slurry while blending. For the composite Coll-I-nHA scaffolds, nano-sized hydroxyapatite particles are synthesized as previously described27 and added to the collagen slurry at a 2:1 ratio to the weight of the collagen during the blending process. All scaffolds are physically crosslinked and sterilized using a dehydrothermal treatment at 105 °C for 24 h25. Cylindrical scaffolds (6 mm diameter, 4 mm height) are obtained using a biopsy punch and can be chemically crosslinked with 3 mM N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride and 5.5 mM N-hydroxysuccinimide (EDAC/NHS) in distilled water (dH2O) to improve the mechanical properties of the constructs30. This well-optimized manufacturing process of two collagen scaffolds creates scaffolds with reproducible mechanical properties, including pore size, porosity, and stiffness (kPa). Both Coll-I-GAG and Coll-I-nHA scaffolds have varying physical properties, creating different environmental conditions. The properties of each scaffold are displayed in Table 1.
| Coll-I-GAG | Coll-I-nHA | |
| Scaffold Size (diameter [mm] x height [mm]) |
6 x 4 17 | 6 x 4 17 |
| Collagen Concentration (wt. %) | 0.5 17 | 0.5 17 |
| Substrate Concentration (wt. %) [based off weight of collagen] |
0.05 15,17 | 200 17 |
| Mean pore size (mm) | 96 22 | 96 – 120 29 |
| Porosity (%) | 99.5 23 | 98.9 – 99.4 27 |
| Stiffness (kPa) | 1.5 27 | 5.5 – 8.63 29 |
Table 1: Overview of the mechanical properties of the two scaffolds adopted for studying neuroblastoma biology.
This paper describes a protocol of assembling a 3D scaffold-based system to better mimic the neuroblastoma microenvironment using neuroblastoma cell lines and previously described collagen-based scaffolds supplemented with either nHA (Coll-I-nHA) or chondroitin-6-sulphate (Coll-I-GAG). The protocol includes downstream methods to analyze the growth mechanisms of the neuroblastoma cells in a more physiologically relevant environment using previously optimized inexpensive methods adapted from 2D monolayer culture Figure 1.
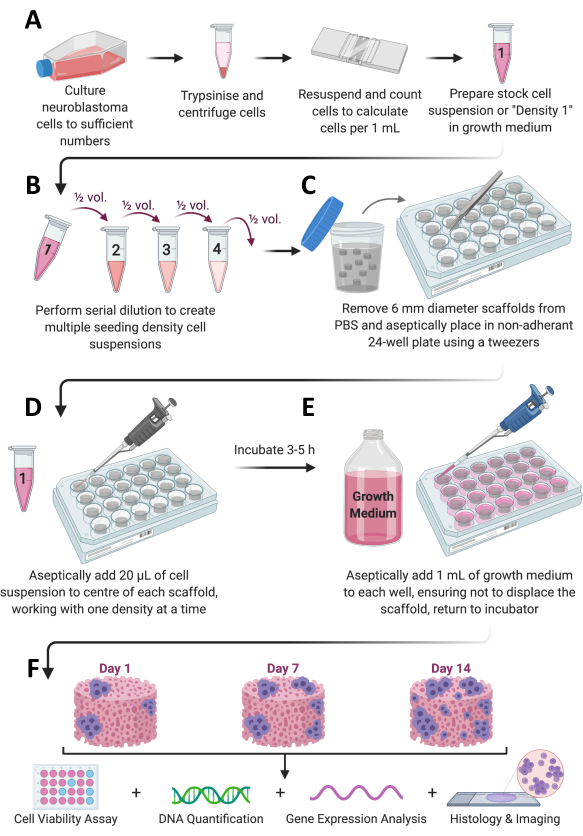
Figure 1: Overall protocol workflow. (A) Cells are grown to sufficient numbers, split, counted, and resuspended in an appropriate volume of medium. (B) This cell stock then undergoes serial dilution to prepare a total of 4 cell suspensions of different densities. (C) Collagen-based scaffolds are sterilely plated in non-adherent 24-well plates, and (D) 20 µL of cell suspension is added to the center of each scaffold and left to incubate at 37 °C, 5% CO2, and 95% humidity for 3-5 h. (E) Complete growth medium (1 mL) is then slowly added to each scaffold, and the plates are placed back into the incubator to allow cell growth for the desired timeframe. (F) At each predetermined time point, several scaffolds are retrieved for cell viability and growth assessment, gene expression analysis, and histological staining. Please click here to view a larger version of this figure.
1. Experimental design
NOTE: The number of scaffolds and cells needed for each experiment will be dependent on the scale of the experiment and can be calculated using the tools in this section on experimental design.
- Number of scaffolds required
- Determine the overall experimental timeline and assessment intervals for changes in cell biology (e.g., cell growth and metabolism).
NOTE: For example, the experimental timeframe is 14 days, with an assessment of cell growth every 7 days. Thus, the experimental time points are Days 1, 7, 14 giving a total of 3 time points. - Next, decide the number of experimental applications to be applied at each time point. Try to complete the same analyses at every time point to monitor the changes.
NOTE: Typical analyses of cell growth on scaffolds include cell viability assays, DNA quantification, histological staining and imaging, and gene expression analysis. These will be further discussed in section 5. - Decide when to use the same scaffold for multiple downstream applications, e.g., after cell viability assessment using an appropriate assay, re-use the scaffold for DNA/RNA isolation (discussed in 5.1.11).
- Keep a minimum of 3 biological replicates for each application, e.g., 3 scaffolds for cell viability and DNA quantification, 3 for histology, and 3 for gene expression analysis. As this equals 9 scaffolds per time point, plan to use 27 scaffolds for 3 time points.
- Finally, multiply this number by the number of experimental parameters or conditions (e.g., assessing multiple cell lines, initial seeding densities, scaffold compositions).
NOTE: In this protocol, 4 different initial seeding densities were being assessed for one cell line, resulting in 108 (27 scaffolds x 4 conditions) scaffolds required. To account for human error and give extra coverage, add ~10% to this number, e.g., if 108 scaffolds are required, prepare 120 scaffolds.
- Determine the overall experimental timeline and assessment intervals for changes in cell biology (e.g., cell growth and metabolism).
- Number of cells required
NOTE: A 20 µL volume of cell suspension is recommended for seeding cells onto a cylindrical scaffold (diameter 6 mm, height 4 mm) on Day 0. The number of cells per this 20 µL of cell suspension is adjusted according to the experimental design, calculated in section 1.1. A common initial seeding density is 2 × 105 cells per 20 µL, used as an example for the below protocol.- To calculate the total amount of cells required for the experiment, multiply the initial 2 × 105 cells per 20 µL by the number of scaffolds required.
NOTE: For example, 30 scaffolds multiplied by 20 µL gives a total volume of 600 µL. If each scaffold requires 2 × 105 cells, 600 µL of suspension contains a total of 6 × 106 cells (2 × 105 × 30), giving a final requirement of 6 × 106 cells in 600 µL. The number of the experimental parameters will dictate the total number of cells required. This protocol, therefore, outlines cell culture using a multilayer cell culture flask, which can handle the same number of cells as 10 traditional 175 cm2 flasks.
- To calculate the total amount of cells required for the experiment, multiply the initial 2 × 105 cells per 20 µL by the number of scaffolds required.
2. Preparation of collagen-based scaffolds
NOTE: Coll-I-nHA and Coll-I-GAG cylindrical scaffolds (diameter 6 mm, height 4 mm) are prepared using established methods15,21,27. Once chemically crosslinked as per previously published methods17, the scaffolds must be used within 1 week.
- Following the manufacture of the scaffolds with the desired mechanical properties, ensure that the scaffolds are fully hydrated and thoroughly washed in phosphate-buffered saline (PBS).
NOTE: This generally takes ~12 h after the crosslinking of the scaffolds and can be carried out in 100 mL tissue culture waste containers at 4 °C, with a maximum of 50 scaffolds per container and 2 mL of PBS per scaffold. - Store the scaffolds in PBS at 4 °C until ready to use.
3. Propagate neuroblastoma cells in a multilayer cell culture flask
NOTE: The optimal seeding density for the multilayer flask will vary. For the flask used in this experiment, the optimal density as per the manufacturer's instructions is 1 × 107 cells. Before seeding the multilayer flask, propagate cells to a density of 1 × 107 cells or higher in an appropriate tissue culture flask (e.g., a T175 cm2 tissue culture flask). To seed cells into the multilayer flask (section 3.1), grow them until 70-80% confluent, harvest, and count the numbers of cells per mL, referring to steps 3.2.16-3.2.20 for performing the cell count. Once the cell suspension is counted, proceed immediately to the seeding of the multilayer flask. Cell culture work must be carried out in a laminar flow hood to maintain sterility.
- Seeding the multilayer cell culture flask
- Prewarm 550 mL of complete growth medium (varies depending on the cell line in use) and 100 mL of sterile PBS in a 37 °C water bath for 20 min.
- Using the harvested cell suspension, calculate the required volume of the cell suspension needed to achieve the optimal seeding density of 1 × 107 cells using equation (1), where WANT refers to the number of cells required for seeding the multilayer flask, and HAVE refers to the number of cells/mL in the cell suspension:
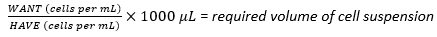 (1)
(1)
e.g.,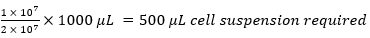
- Add the required volume of the cell suspension to 100 mL of the prewarmed growth medium.
- Take a new multilayer flask in the hood, remove the cap, and hold the flask at a 60° angle. Slowly pipette all 100 mL of the cell suspension into the flask, down the angled side of the neck. Cap the flask and place it on its side to allow the cells to distribute throughout the layers evenly.
- At a 60° angle, add 400 mL of the prewarmed growth medium to the multilayer flask by slowly pouring or pipetting down the angled side of the neck. If the neck becomes full, return the flask to an upright position, or cap the flask and place it on its side before returning to pouring.
NOTE: Pour slowly to avoid excessive bubble formation. Gently tap the flask in the upright position to allow all bubbles to rise to the top and remove them with a 10 mL pipette. Ensure the flask is filled to the bottom thread of the neck; add more medium, if necessary, to achieve this. - Cap the multilayer flask and incubate it with the angled neck facing down at 37 °C, 5% CO2, and 95% humidity.
- Check the growth every 2-3 days for confluency. To check the confluency of the bottom two layers of the multilayer flask, view them under a 4x objective lens of an inverted microscope.
NOTE: When seeded with 1 × 107 neuroblastoma cells, the 10-layer flask will typically take one week to become confluent-though this may vary depending on the cell line used.
- Routine maintenance of cells in the multilayer flask
- Prewarm 550 mL of complete growth medium (varies depending on the cell line in use), 50-100 mL of trypsin, and 300 mL of sterile PBS in a 37 °C water bath for 20 min.
- Check the multilayer flask for 70-80% confluency.
- Place the multilayer flask in a laminar flow hood and discard the spent medium from the flask by pouring. Initially, tilt the flask so that the medium is pouring over the air dam into a waste container. While pouring, rotate the flask by 180° until the medium is flowing down the angled neck of the flask. Rotate the flask back and forth along this axis to eliminate any remaining liquid.
- Wash the cells by adding 100 mL of prewarmed sterile PBS slowly down the angled neck. Cap the flask, place it on its side to allow uniform distribution of PBS, and rotate the flask back and forward to wash the cells.
- Discard the PBS wash in the same manner as 3.2.3. Repeat the washing steps.
- Dilute 50 mL of the prewarmed trypsin in 50 mL of prewarmed sterile PBS. Add 100 mL of diluted trypsin solution to the multilayer flask, cap, and place the flask on its side to allow uniform distribution of the trypsin. If the cell line is highly adherent, use 100 mL of undiluted trypsin.
- Incubate the flask for 2-5 min at 37 °C, 5% CO2, and 95% humidity, monitoring cell detachment under the microscope. If necessary, tap the flask firmly to aid detachment or place it back in the incubator for one more minute.
- Place the multilayer flask in a laminar flow hood and neutralize the trypsin with 100 mL of growth medium. Cap the flask, place it on its side, and rock back and forward to ensure complete neutralization.
- Pour off the neutralized cell suspension into 4 x 50 mL conical centrifuge tubes.
NOTE: If complete cell harvest is required, wash the multilayer flask again with 100 mL of sterile PBS and pour it off into 2 x 50 mL centrifuge tubes. - Centrifuge the cell suspension in 50 mL centrifuge tubes at 340 × g for 3-4 min to pellet the cells.
- Return the centrifuge tubes to the laminar flow hood and carefully discard as much supernatant as possible from each pellet.
NOTE: The pellet will be large and relatively loose and hence, can be disrupted easily. - Add 1-5 mL of growth medium to each pellet and resuspend it by pipetting up and down several times.
- Pool together the 4 resuspended pellets in one 50 mL centrifuge tube, mix them thoroughly with the pipette, and note the total volume.
- Make an appropriate dilution of the cell suspension for cell counting so that each outer square on the hemocytometer contains 30-100 cells.
NOTE: A suitable starting dilution is 1:100; for this, pipette 10 µL of the cell suspension into a fresh 15 mL conical centrifuge tube and dilute it with 990 µL of sterile PBS. Pipette the mixture up and down several times to mix thoroughly. - Place a 50 mL centrifuge tube containing the cell stock in the incubator while counting.
- Taking a clean hemocytometer, place a clean coverslip over the grid, as shown in Figure 2.
- Pipette 10 µL of the diluted cell suspension into the hemocytometer chamber. If the chamber becomes full before all of the 10 µL is dispensed, stop pipetting.
- Place the hemocytometer under the 4x objective of a light microscope. Adjust the coarse and fine focus to visualize the cells.
- Count the number of cells in the four outer corner squares of the chamber as highlighted in Figure 2. Add the four counts and divide by 4 to calculate the average cells per square.
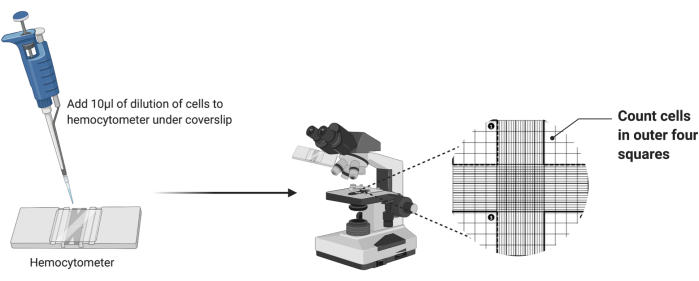
Figure 2: Cell counting using a hemocytometer. Ten microliters of cell suspension are added to the hemocytometer beneath the coverslip. The chamber is then placed under the 4x objective lens of a microscope, and the number of cells in the four outer corners of the grid are counted. Please click here to view a larger version of this figure.
- Multiply the average count by the dilution factor (e.g., 100) and multiply this number by 10,000 to obtain the number of cells per mL using equation (2).
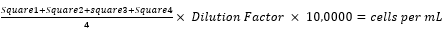 (2)
(2) - Calculate the total number of cells in the stock solution by multiplying by the total volume of the cell stock (e.g., if the 4 resuspended pellets pooled made a 20 mL solution, multiply cells/mL by 20).
- To maintain the multilayer flask, use equation (1) outlined in step 3.1.2 to calculate the volume of cell stock needed to seed 1 × 107 cells back into the flask and carry out steps 3.1.3-3.1.6 to re-seed the flask. If ready to seed the scaffolds, proceed to the next section.
4. Seed neuroblastoma cells on scaffolds
- Prepare stock cell suspension
NOTE: This protocol will outline the steps for creating four different seeding densities of neuroblastoma cells, with a multiplication factor of 2 between each density. A serial dilution will therefore be used to create three further cell suspensions from the stock suspension. Cell culture work must be carried out in a laminar flow hood to maintain sterility.- Use equation (3) to calculate the volume of cells needed from the total number of cells in the multilayer flask (counted in section 3.2.19) to prepare the first seeding density or cell stock suspension.
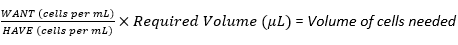 (3)
(3)
NOTE: For example, if the highest seeding density is 6 × 105 cells per scaffold required for 30 scaffolds, with each scaffold receiving 20 µL of cell suspension, the stock cell suspension would require 1.8 × 107 cells (6 × 105 cells × 30 scaffolds) in a total volume of 600 µL (20 µL × 30 scaffolds).
As a serial dilution will be performed from this preparation, these numbers must be doubled, i.e., 3.6 × 107 cells in a total volume of 1200 µL. To convert this to WANT in cells per mL; divide 3.6 × 107 by 1200 µL and multiply by 1000 µL, giving 3 × 107 cells per mL.
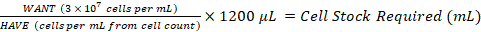
- Add the required volume of cell stock to a sterile 2 mL or 15 mL centrifuge tube and bring to a final volume of 1200 µL with growth medium. Label this tube as Density 1 (Figure 3).
- Use equation (3) to calculate the volume of cells needed from the total number of cells in the multilayer flask (counted in section 3.2.19) to prepare the first seeding density or cell stock suspension.
- Perform serial dilution to create multiple seeding density cell suspensions.
- De Density 1 prepared in step 4.1.1, prepare three more densities of cell suspension through serial dilution as per Figure 3.
- Dilute each density by a factor of 2 in the growth medium. First, add the required final volume (600 µL from the previous example) of growth medium to three sterile 2 mL or 15 mL centrifuge tubes.
- Transfer half of Density 1 (600 µL) to one of the tubes, thoroughly mixing the cell suspension with the medium to dilute. Label this tube as Density 2.
- Transfer half of Density 2 (600 µL) to the next tube, thoroughly mixing the cell suspension with the medium to dilute. Label this tube as Density 3.
- Transfer half of Density 3 (600 µL) to the next tube, thoroughly mixing the cell suspension with the medium to dilute. Label this tube as Density 4.
- Discard 600 µL from Density 4 so that all four preparations have a final volume of 600 µL.
- As a negative control, add 600 µL of growth medium only to a sterile 2 mL centrifuge tube. See Figure 3 for a schematic of the serial dilution process.
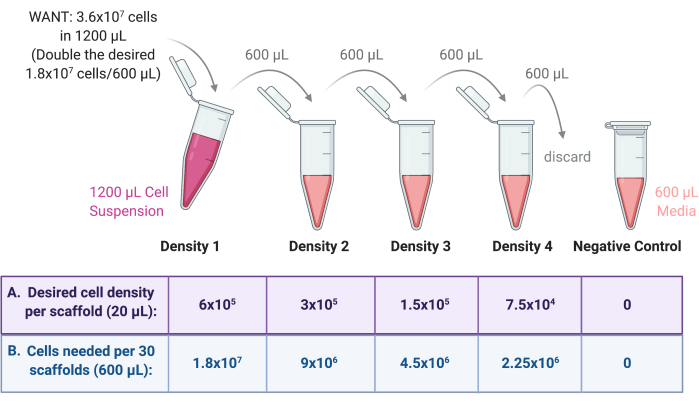
Figure 3: Serial dilution of cell stock to prepare 4 suspensions for 4 different scaffold seeding densities. (A) Numbers can be adjusted to suit the desired seeding density per scaffold and (B) multiplied for the total number of scaffolds per density, with each scaffold receiving 20 µL of cell suspension. In this example, Density 1 requires 6 × 105 cells per scaffold, equivalent to 1.8 × 107 cells in 600 µL for 30 scaffolds. This number is doubled to begin the serial dilution, as 600 µL is then transferred and diluted in 600 µL of growth medium in the next tube. This process continues until there are 4 cell suspensions with a factor of 2 between each. A negative control is made by adding 600 µL of medium only to a tube. Please click here to view a larger version of this figure.
- Adding cell suspensions to the scaffolds
NOTE: Remove the scaffolds (stored in PBS) from the refrigerator and allow them to come to room temperature (RT) before adding the cells.- Bring the scaffolds in PBS into the laminar flow hood.
- Using sterile tweezers, place the scaffolds in non-adherent 24-well plates with one scaffold per well (Figure 1C). Gently lift the scaffolds by their corners and lightly press them against the side of the container to remove excess PBS. Add the scaffolds into the center of the wells skin-side-down (the shiny layer side of the scaffold, facing down into the plastic 24-well plates).
- Label the 24-well plates with details of the cell line, relevant parameters (e.g., seeding density), and time points. Work with one cell seeding density at a time, keeping the remaining densities in the 37 °C incubator until ready for use.
- In the laminar flow hood, use a P20 pipette and sterile tips to gently add 20 µL of the relevant cell suspension to the center of each scaffold (Figure 1D). Keep the cells thoroughly in suspension by mixing well while adding the cells to the scaffolds. Ensure that the suspension remains on top of the scaffold and does not slide off onto the base of the well, as this will not allow cell attachment to the scaffold.
- Once the cells have been added, incubate the plates for 3-5 h (37 °C, 5% CO2, and 95% humidity) to allow most of the cells to attach.
- After incubation, slowly and gently add 1 mL of prewarmed growth medium to each well (Figure 1E). Use a P1000 pipette to add the medium to allow for more slow and controlled motion, preventing displacement of the scaffolds. If working with a very high number of scaffolds, use a 10 mL pipette with the pipette gun set to 'drop' and 'low.'
- Incubate the 24-well plates overnight (37 °C, 5% CO2, and 95% humidity).
- Maintenance of cells on the scaffolds
- After the first 24 h of cell attachment (Day 1), transfer the seeded scaffolds to new non-adherent 24-well plates, and add 1-2 mL of fresh growth medium.
NOTE: This step removes cells that have fallen to the bottom of the plastic 24-well plates rather than allowing them to grow on the scaffolds. Scaffold replicates designated as Day 1 will be taken down after 24 h, as discussed in section 5; hence, maintenance does not apply to these scaffolds. - Monitor the scaffolds initially every 2-3 days for a change in color of the growth medium. As time progresses and cells proliferate within the scaffolds, feed the cells more frequently.
- Using a 10 mL pipette gun, on slow mode, remove the 1 mL of the spent medium and discard. If carrying out experiments requiring conditioned medium, collect the spent medium of biological replicates in a 15 mL centrifuge tube, centrifuge at 340 × g for 2 min to pellet the cellular debris, transfer the supernatant to a fresh tube, and store at -80 °C.
- Gently add 2 mL of prewarmed growth medium to the scaffolds, using the drip function again on the pipette, and return the 24-well plate to the incubator (37 °C, 5% CO2, and 95% humidity). Repeat whenever the medium is spent for the duration of the desired growth period.
- After the first 24 h of cell attachment (Day 1), transfer the seeded scaffolds to new non-adherent 24-well plates, and add 1-2 mL of fresh growth medium.
5. Scaffold retrieval and applications
NOTE: At each time point, several applications can be used to monitor cell growth on the scaffolds or assess gene and protein expression profiles. The conditions of scaffold retrieval will depend on the analysis to be performed, with multiple retrieval methods outlined in the following subsections and demonstrated in Figure 4.
- Assessment of cell viability within scaffolds
- Sterilize the appropriate cell viability assay reagent by filtering through a 0.2 µm sterile filter into a centrifuge tube in the laminar flow hood. Prewarm this sterile solution along with complete growth medium and sterile PBS in a 37 °C water bath.
- In the laminar flow hood, use sterile tweezers to transfer the scaffolds to be analyzed to a fresh 24-well plate. Label the plate with all relevant details.
NOTE: Perform the analysis in triplicate. - Add 900 µL of the prewarmed growth medium to each well, followed by 100 µL of the sterile cell viability reagent. Include a negative control by adding 900 µL of medium and 100 µL of the sterile cell viability reagent to a well with no scaffold. Replace the lid on the plate, and gently rock the plate for ~3 min to evenly distribute the diluted cell viability reagent throughout the well. Incubate the plate at 37 °C, 5% CO2, and 95% humidity.
NOTE: Incubation times will need to be optimized for each cell line; refer to the manufacturer's guidelines. For neuroblastoma cell lines, 4-6 h incubation appears optimal. - After incubation, remove the plate from the incubator and gently rock it for a few seconds.
- In the laminar flow hood, open a new, translucent 96-well plate. From each well in the 24-well plate, transfer the incubated medium and reagent to three wells of the 96-well plate with 100 µL per well, giving technical triplicates.
NOTE: This transfer will leave 700 µL in the wells of the 24-well plate. - Cover the 96-well plate in aluminum foil to protect the cell viability reagent from light.
- Remove and discard the remaining 700 µL of the well contents from each scaffold in the 24-well plate. Wash each scaffold twice with 1 mL of sterile PBS.
NOTE: All color will not be removed from the scaffolds. These scaffolds can then be used for further applications, e.g., DNA quantification, by placing them in 1 mL of 1% Triton X-100 in 0.1 M sodium bicarbonate (NaHCO3) solution and storing them at -80 °C (see section 5.2, Figure 4B). - Remove the 96-well plate from the laminar flow hood and measure the absorbance of each well at wavelengths of 570 nm and 600 nm using a microplate reader. Record the absorbance values at both wavelengths and follow the manufacturer's instructions to calculate the percentage reduction of the cell viability reagent by the cells.
- Graph and statistically analyze the cell viability results using appropriate software. Input biological triplicate values to produce error bars and indicate the assay variability.
- To examine changes in cell viability over the experimental timeframe, perform a one-way analysis of variance (ANOVA) test with multiple comparisons of the means using appropriate biostatistical software.
- Indicate significant differences between the time points on graphs as ns (P>0.05), * (P≤0.05), ** (P≤0.01), *** (P≤0.001), and **** (P≤0.0001).
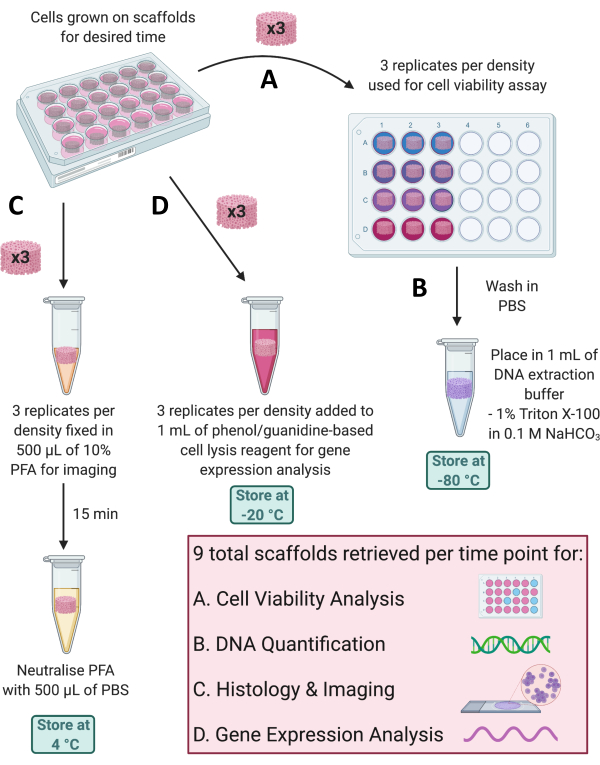
Figure 4: Retrieval of scaffolds for different analyses at each time point. (A) Three scaffold replicates are retrieved for cell viability analysis. (B) These scaffolds can then be washed in PBS, placed in 1% Triton X-100 in 0.1 M NaHCO3, and stored at -80 °C for DNA quantification. (C) Three more replicates are fixed in 10% PFA for 15 min, neutralized in PBS, and stored at 4 °C for histological staining and imaging. (D) Finally, 3 replicates are added to a phenol/guanidine-based cell lysis reagent and stored at -20 °C for gene expression analysis. Abbreviations: PBS = phosphate-buffered saline; PFA = paraformaldehyde. Please click here to view a larger version of this figure.
- Quantification of DNA from the cells in the scaffolds
NOTE: As mentioned in the note after step 5.1.7, the retrieval of the scaffolds for DNA quantification involves placing the scaffolds into 2 mL centrifuge tubes containing 1 mL of 1% Triton X-100 in 0.1 M NaHCO3 solution followed by storage at -80 °C. Before DNA analysis can be performed, the cells must undergo three freeze-thaw cycles to lyse the neuroblastoma cells appropriately and release DNA for quantification.- Remove the samples previously stored in Triton X-100 from -80 °C. Leave the samples at RT for 1-3 h or until thawed.
- Vortex the samples for 10-20 s, and place the samples back at -80 °C for 18-24 h or until completely frozen. Repeat this process for a total of three freeze-thaw cycles.
- To maximize the DNA yield, use a tissue lyser to disrupt the cells in the scaffolds.
- Place a metal bead in the 2 mL centrifuge tube containing a scaffold in Triton X-100, and place the tube within the adaptor to shake the sample at 50 oscillations/second for 2-3 min.
NOTE: Use round-bottom centrifuge tubes as the metal bead may become lodged in a tapered-bottom tube.
- Place a metal bead in the 2 mL centrifuge tube containing a scaffold in Triton X-100, and place the tube within the adaptor to shake the sample at 50 oscillations/second for 2-3 min.
- Quantify the DNA in the Triton X-100 solution by applying a fluorescent double-stranded DNA (dsDNA) stain and measure the emission using a microplate reader. Refer to the manufacturer's guidelines; dilute the samples appropriately in Tris-ethylenediamine tetraacetic acid (TE) buffer to prepare 8 dsDNA standards through serial dilution in TE buffer (Figure 5).
- In black opaque 96-well plates, add 100 µL of each standard or sample to the wells in technical triplicate.
- Dilute the fluorescent dsDNA stain 200-fold in TE buffer and add 100 µL to each standard/sample using a multichannel pipette. Cover the plate in tinfoil and incubate at RT for 5 min.
- Measure and record the fluorescence of each well at 520 nm. Follow the manufacturer's guidelines to calculate the concentration of DNA in each sample.
NOTE: If the average concentration of DNA per cell is known for the cell line being used, DNA concentration values can be converted to cell numbers using equation (4).
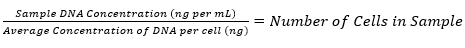 (4)
(4) - Graph and statistically analyze the DNA quantification results using appropriate software. Input biological triplicate values to produce error bars and indicate the assay variability.
- To examine the changes in DNA concentration/cell numbers over the experimental timeframe, perform a one-way ANOVA test with multiple comparisons of the means using appropriate biostatistical software.
- Indicate significant differences between time points on graphs as ns (P>0.05), * (P≤0.05), ** (P≤0.01), *** (P≤0.001), and **** (P≤0.0001).
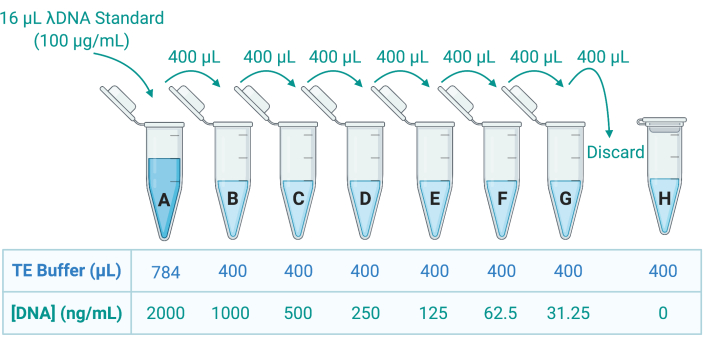
Figure 5: Preparation of eight DNA standards for the generation of a standard curve. A stock solution of λDNA is provided at 100 µg/mL. This is diluted 50-fold in TE buffer to create standard A at 2000 ng/mL; 400 µL of A is then transferred to tube B, containing 400 µL TE buffer; 400 µL of B is then transferred and diluted 2-fold in C, and so on until G. Standard H is composed of only TE buffer and therefore has a DNA concentration of 0 ng/mL. Abbreviation: TE = Tris-EDTA. Please click here to view a larger version of this figure.
- Preparation of scaffolds for histological staining
NOTE: Scaffolds can be fixed and stained for microscopy and imaging purposes either as whole scaffolds for immunofluorescence (IF) or as formalin-fixed paraffin-embedded (FFPE) slices for histological staining or immunohistochemistry (IHC). This allows the qualitative assessment of cell penetration and distribution within scaffolds and can be used to assess the expression of proteins.- Prepare a 10% paraformaldehyde (PFA) solution in PBS. Ensure that there is enough solution for a final volume of 500 µL per scaffold.
- Prewarm this solution at 37 °C and add 500 µL to labeled 2 mL centrifuge tubes for scaffold retrieval.
- Remove the scaffolds from the non-adherent 24-well plates (step 4.4.4) and place them in the laminar flow hood.
- Using sterile tweezers, transfer the scaffolds to the labeled centrifuge tubes containing 10% PFA. Allow the scaffolds to fix in the PFA solution for 15 min. Neutralize the PFA by adding 500 µL PBS to each tube and store at 4 °C.
- To prepare the scaffolds for the Automatic Tissue Processor, remove them from 4 °C and use tweezers to place them in plastic cassettes labelled with all relevant details in pencil. Place all the cassettes in the metal container of the Tissue Processor.
- Begin the 12-stage protocol on the Tissue Processor to fix, dehydrate, clear the scaffolds, and infiltrate them with paraffin overnight. Collect the cassettes containing the processed samples.
- Next, embed the scaffolds in paraffin wax blocks to allow microtomy of the scaffolds into very thin slices for staining.
NOTE: It is especially important to consider the orientation of the scaffolds when removing them from the cassettes and embedding them in wax, as this will affect the angle at which the images are taken. This is important when assessing cell infiltration into the scaffolds. - Turn on the wax embedder and the cold plate; lift the lid to check the level of the wax. Refill if necessary.
- Working with one sample at a time, open the cassette, remove the scaffold, and center it in the plastic mold.
- Pour hot wax onto the sample, ensuring that correct orientation is maintained and adjusting with warm tweezers, if necessary, before the wax solidifies. Pour more wax to fill the mold.
- Place the labeled cassette lid on top of the plastic mold and add wax on top. Place the mold on the cold plate to solidify the wax. Store overnight at 4 °C to ensure that the paraffin wax is fully solidified before microtomy.
- To prepare for microtomy, turn on a 35 °C water bath, the drying plate, and the microtome.
- Insert a blade in the holder and tighten the lever to secure it.
- Set the trim and section thickness, generally 5 mm for scaffold sections.
- Remove the FFPE scaffold from the mold, secure it in the holder on the front of the microtome, and carefully trim the excess wax around the edges of the sample before cutting sections.
- Start cutting into the wax block by rotating the lever of the microtome, ensuring smooth motion.
- Collect ribbon-like sections, around 3 scaffold sections at a time, and gently place them in the 35 °C water bath to remove wrinkles. Gently separate the sections while in the water bath using tweezers.
- Using polysine-coated glass microscope slides, lift each section from the water bath so that the section sits in the center of the slide. Label each slide with a pencil.
- Place the glass slides on the drying plate or in a 60 °C drying oven. Once dried, store them at 4 °C and proceed with the required histological or IHC stain.
- Retrieval of scaffolds for gene expression analysis
- Remove the scaffolds from the non-adherent 24-well plates from the incubator (step 4.4.4) and place them in the laminar flow hood.
- Using sterile tweezers, transfer the scaffolds to fresh labeled 2 mL centrifuge tubes.
- In a fume hood, add 1 mL of a phenol/guanidine-based cell lysis reagent to each tube to lyse the cells in scaffolds and allow for recovery of high-quality RNA.
- Store tubes at -20 °C until ready to perform RNA extraction using an appropriate kit. Using a standard reverse-transcription quantitative polymerase chain reaction (RT-qPCR)17, assess gene expression in the cells in the scaffolds.
The collagen-based scaffold model described here has many applications ranging from studying neuroblastoma biology to the screening of anticancer therapeutics in an environment that is more physiologically similar to native tumors than conventional 2D cell culture. Before testing a given research question, it is crucial to obtain a complete characterization of cell attachment, proliferation, and infiltration within the desired experimental timeframe. The growth conditions will depend on the biology of each specific cell line. Importantly, several methods of cell growth assessment must be implemented to determine optimal conditions and robust performance.
Here, the viability of neuroblastoma cells grown on scaffolds was assessed using a colorimetric cell viability assay. This assay can be performed as frequently as desired throughout the experimental timeframe. For the described experiment, cell viability assessment was performed on days 1, 7, and 14 for two neuroblastoma cell lines, KellyLuc and IMR32, grown on Coll-I-nHA scaffolds at 4 different densities (Figure 6). Viability on Day 1 was set as a baseline to compare all subsequent measurements. The rate of reduction of the cell viability reagent is reflective of the cell biology and growth characteristics of individual cell lines, including their proliferation rates and metabolism. A correlation between the number of cells seeded on the scaffolds and the level of reduction was expected. In this experiment, the reduction of the cell viability reagent generally increased with each time point for both cell lines at all densities, as expected.
Each density was then assessed individually for both cell lines to compare the reduction across time points. One-way ANOVA with Tukey's multiple comparisons test was performed to detect significant differences in reduction between time points (Figure 7). For both cell lines and all seeding densities, there was a significant increase (P<0.05) in the reduction of the cell viability reagent when comparing day 1 and day 14. This indicated a significant increase in metabolically active cells present on the scaffolds. This increase was not significant in all cases when assessing the 7-day intervals (day 1 vs. day 7, day 7 vs. day 14), demonstrating the importance of the optimization of the seeding density to achieve the desired growth window.
To support the results of the cell viability assay, cell growth on scaffolds can also be indirectly measured via the quantification of dsDNA extracted from scaffolds using a fluorescent dsDNA stain (Figure 8A). Like cell viability, DNA quantification can be done as frequently as desired within the experimental timeline. However, this analysis requires the complete retrieval of scaffolds and termination of cell growth and so must be factored into experimental planning as discussed in section 1. For this experiment, DNA was quantified on days 1, 7, and 14 for two neuroblastoma cell lines, KellyLuc and IMR32, grown on Coll-I-nHA scaffolds at 4 different densities. As the average concentration of dsDNA per cell is known for these cell lines, it was possible to derive the number of cells per sample from the quantified DNA (Figure 8B).
DNA quantification gave rise to higher variability between biological replicates than cell viability assessment but generally increased for each time point, with the highest levels quantified on day 14. IMR32 cells appear to reach higher cell numbers on Coll-I-nHA scaffolds, as indicated by DNA concentration, than KellyLuc cells. Each density was then assessed individually for the two cell lines to compare the reduction across time points. One-way ANOVA with Tukey's multiple comparisons test was performed to detect significant differences in reduction between time points (Figure 8B).
For both cell lines and all seeding densities, there was a significant increase (P<0.05) in cell numbers when comparing day 1 and day 14, with the exception of KellyLuc at seeding density 4 (1 × 105 cells/scaffold), which did not yield significant increases across any of the time points. Similar to the cell viability results, the increases were not significant in all cases when assessing the 7-day intervals (day 1 vs. day 7, day 7 vs. day 14). When comparing the time point trends for cell viability and DNA quantification, there were some slight differences between the two analyses. However, overall similar trends were observed, with mean values increasing between 7-day intervals for most densities. This demonstrates the importance of monitoring cell growth using more than one method.
A visual assessment of cell growth morphology and distribution on the scaffolds was next implemented, encompassing traditional hematoxylin and eosin (H&E) staining as well as IHC. It is expected that the different growth patterns of individual cell lines will lead to varied spatial arrangements on scaffolds, including different degrees of penetration into the scaffold and cell clustering. Scaffolds were formalin-fixed, paraffin-embedded, and cut into 5 mm sections (Figure 9A), preparing the scaffolds for multiple visualization techniques, including histological staining and IHC.
Routine H&E staining was applied to Kelly, KellyCis83, and IMR32 cells grown on collagen-based scaffolds on days 1, 7, and 14 (Figure 9B). This allowed visualization of the cells' spatial orientation on two collagen-based scaffolds over a 14-day period. Cisplatin-sensitive Kelly cells and resistant KellyCis83 cells were grown on both Coll-I-nHA scaffolds (Figure 9B, i) and Coll-I-GAG scaffolds (Figure 9B, ii). Consistent with previously published data, KellyCis83 cells grew at a higher rate and infiltrated deeper into both scaffold compositions than the less invasive Kelly cell line. The H&E stain of another neuroblastoma cell line, IMR32, grown on Coll-I-nHA demonstrates a contrasting growth pattern (Figure 9B, iii). This cell line grew in large, densely packed clusters on the collagen scaffolds over the 14-day period. Brightfield confocal microscopy can be used to visualize the porous architecture of collagen-based scaffolds (Figure 9C) owing to the autofluorescence of collagen fibers.
We stained cells with phalloidin targeting cytoskeletal actin and the nuclear counterstain, 4′,6-diamidino-2-phenylindole (DAPI), to monitor specific cell traits throughout the experimental timeline. An abundance of actin was observed in Kelly and KellyCis83 cells on Coll-I-GAG scaffolds using this technique (Figure 9D). These results demonstrate how multiple imaging techniques can be used to derive spatially resolved information from neuroblastoma cells grown on scaffolds using this protocol. This characterization of cell growth patterns on collagen-based scaffolds over a given period will improve the understanding and interpretation of any downstream biochemical assays.
Protein expression by cells grown on collagen-based scaffolds can be analyzed to compare cellular activity to in vivo scenarios. Previously published data examined the expression of chromogranin A (CgA) as a surrogate secreted marker of neuroblastoma by KellyLuc and KellyCis83Luc cells grown in cell monolayers as well as on Coll-I-nHA and Coll-I-GAG scaffolds (Figure 10). CgA was assessed in the conditioned media using an enzyme-linked immunosorbent assay (ELISA) (Figure 10A). CgA is secreted at a higher rate in the more aggressive chemo-resistant KellyCis83 cell line than in Kelly (Figure 10B,C). This was significant on day 7 on both Coll-I-GAG and Coll-I-nHA scaffolds (P<0.05), whereas there was no significant difference at this time point for cells grown as a monolayer by conventional 2D culture.
These results also highlight the restricted experimental timeline when growing cells in a monolayer, with only 7 days of growth proving feasible before cells reach confluency. The growth of cells on scaffolds overcomes this limitation as they can be maintained over a longer period in more physiologically relevant conditions. The above combination of techniques to acquire information on cell viability, DNA content, cellular morphology and spatial arrangement, and expression profiles facilitates the assessment of the growth of neuroblastoma cells on a range of collagen-based scaffolds. This protocol can also be easily adapted to satisfy specific experimental requirements and desired applications.
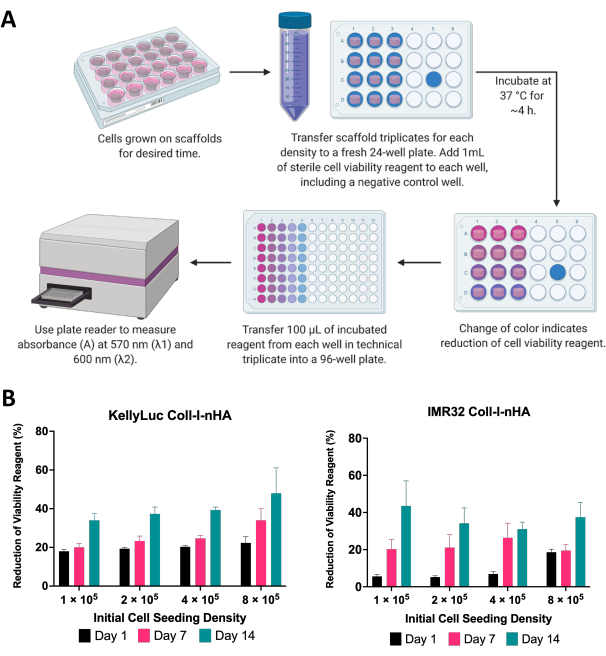
Figure 6: Cell viability analysis. (A) General procedure for measuring the viability of neuroblastoma cells on collagen-based scaffolds using a colorimetric cell viability assay. The incubation period must be optimized for each new cell line, referring to the manufacturer's guidelines. (B) Percentage reduction of cell viability reagent by KellyLuc and IMR32 cells grown on Coll-I-nHA scaffolds at four different initial seeding densities, measured on days 1, 7, and 14. Samples were assessed in biological triplicate with error bars representing the standard deviation. Abbreviations: nHA = nanohydroxyapatite; Coll-I-nHA = collagen scaffolds supplemented with nHA. Please click here to view a larger version of this figure.
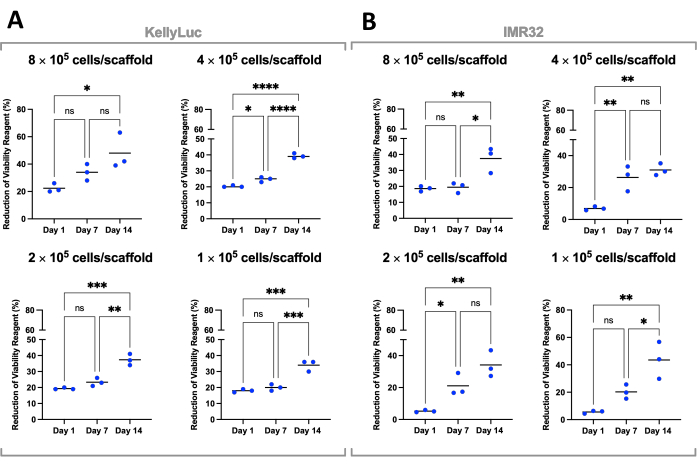
Figure 7: Cell viability by seeding density for cells grown on Coll-I-nHA over a 14-day period. (A) KellyLuc;(B) IMR32.Titled cell numbers refer to the initial cell seeding density on the scaffolds on Day 0. Samples were assessed in biological triplicate, indicated by triplicate points, with bars representing the mean. One-Way ANOVA with Multiple Comparisons was used to detect significant differences in % cell viability reagent reduction across the three time points, noted on the graphs (ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001). Abbreviations: nHA = nanohydroxyapatite; Coll-I-nHA = collagen scaffolds supplemented with nHA; ANOVA = analysis of variance; ns = not significant. Please click here to view a larger version of this figure.
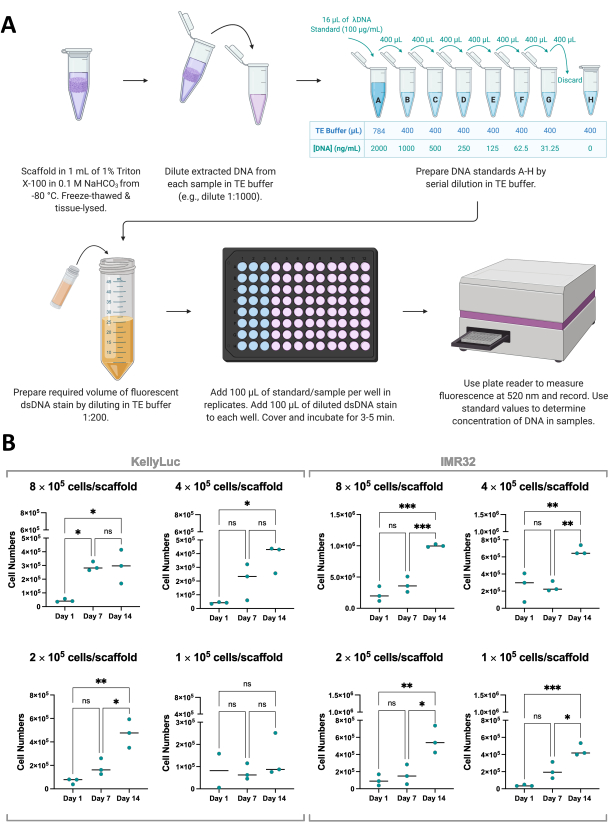
Figure 8: Quantification of DNA extracted from cells in scaffolds. (A) Process of quantifying dsDNA from cells grown on collagen-based scaffolds using a fluorescent dsDNA stain. (B) Cell numbers from DNA quantification analysis by seeding density for KellyLuc and IMR32 cells grown on Coll-I-nHA over a 14-day period. Titled cell numbers refer to the initial cell seeding density onto scaffolds on Day 0. Samples were assessed in biological triplicate, indicated by triplicate points, with bars representing the mean. One-Way ANOVA with Multiple Comparisons was used to detect significant differences in cell numbers across the three time points, noted on the graphs (ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001). Abbreviations: nHA = nanohydroxyapatite; Coll-I-nHA = collagen scaffolds supplemented with nHA; dsDNA = double-stranded DNA; TE = Tris-EDTA; ANOVA = analysis of variance; ns = not significant. Please click here to view a larger version of this figure.
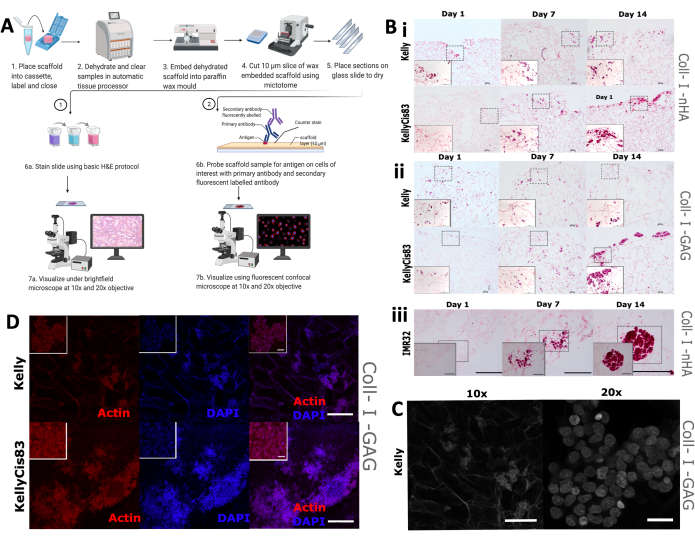
Figure 9: Tissue processing steps for immunohistochemistry analysis of scaffolds. (A) Schematic representation of the protocol for processing scaffolds for image analysis. This process allows routine histological staining and specific antibody probing using primary antibodies and fluorescently labeled secondary antibodies. (B) Representative images of three neuroblastoma cell lines subjected to H&E staining. H&E images are taken on Days 1, 7, 14 to monitor growth patterns over the time course of the experiment. Scale bar = 200 µm. Dashed squares represent the area that was chosen for zoomed-in 20x images at the lower left edge. Scale bar = 20 µm. (i and ii) H&E of Kelly and KellyCis83 neuroblastoma cell lines (upper and lower panels, respectively) on two types of collagen-based scaffolds. (iii) H&E of IMR32 cell line, representing clustered cellular growth on the Coll-I-nHA scaffold. (C) Representative image of the Kelly cell line, subjected to brightfield confocal microscopy. The collagen autofluorescence allows visualization of the porous scaffold. 10x Scale bar = 200 µm, 20x scale bar = 20 µm. (D) Representative image of embedded scaffolds followed by analysis by IHC with phalloidin and DAPI at 10x magnification, Scale bar = 200 µm. Smaller inside squares represent zoomed-in images (20x), scale bar = 20 µm. Abbreviations: nHA = nanohydroxyapatite; Coll-I-nHA = collagen scaffolds supplemented with nHA; GAG = glycosaminoglycan; Coll-I-GAG = collagen scaffolds supplemented with chondroitin-6-sulfate; H&E = hematoxylin and eosin; IHC = immunohistochemistry; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
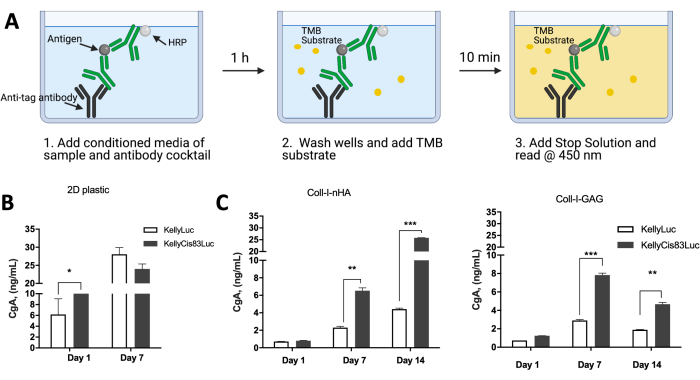
Figure 10: Protein expression by neuroblastoma cells grown on 3D collagen-based scaffolds compared to 2D plastic. (A) A schematic of how the CgA ELISA was performed on conditioned media of cells grown on 2D plastic or 3D collagen-based scaffolds. (B) CgA protein expression levels taken from conditioned media of cells grown on a 2D plastic monolayer. As the cells reached confluency after 7 days, the 14-day time point was not readable. By day 7 on plastic, there was no significant difference in CgA levels between Kelly and KellyCis83 cell lines. (C) CgA ELISA performed using conditioned media of cells grown on collagen-based scaffolds for 14 consecutive days. On day 7, on both collagen scaffolds, CgA levels are higher in the more aggressive KellyCis83 cell line, highlighting more physiological relevant levels of CgA in 3D matrix compared to 2D monolayer. This figure has been modified from Curtin et al.17. Abbreviations: 3D = three-dimensional; 2D = two-dimensional; CgA = chromogranin A; ELISA = enzyme-linked immunosorbent assay; nHA = nanohydroxyapatite; Coll-I-nHA = collagen scaffolds supplemented with nHA; GAG = glycosaminoglycan; Coll-I-GAG = collagen scaffolds supplemented with chondroitin-6-sulfate; TMB = 3,3',5,5'-tetramethylbenzidine; HRP = horseradish peroxidase. Please click here to view a larger version of this figure.
