RNA-seqは、通常、差次的遺伝子発現の推定および遺伝子発見1に広く使用されてきました。また、異なるアイソフォームを発現する遺伝子によるエクソンレベルの使用状況の推定にも利用でき、転写後レベルでの遺伝子制御の理解に貢献します。真核生物遺伝子の大部分は、選択的スプライシング(AS)によって異なるアイソフォームを生成し、mRNA発現の多様性を高めます。ASイベントは、異なるパターンに分けることができる:完全エクソン(SE)のスキップ(カセット)エクソンが、その隣接するイントロンと共に転写物から完全に除去される。エクソンの両端に2つ以上のスプライス部位が存在する場合の代替(ドナー)5’スプライス部位選択(A5SS)および代替3′(アクセプター)スプライス部位選択(A3SS);イントロンが成熟mRNA転写物内に保持される場合のイントロンの保持(RI)、および一度に2つの利用可能なエクソンのうちの1つだけを保持できるエクソン使用の相互排除(MXE)2,3。代替ポリアデニル化(APA)はまた、単一の転写産物から複数のmRNAアイソフォームを生成する代替ポリ(A)部位を用いて遺伝子発現を調節する上で重要な役割を果たす4。ほとんどのポリアデニル化部位(pA)は3’非翻訳領域(3′ UTR)に位置し、多様な3′ UTR長のmRNAアイソフォームを生成します。3′ UTRは調節要素を認識するための中心的なハブであるため、異なる3′ UTR長はmRNAの局在、安定性および翻訳に影響を与える可能性があります5。プロトコル6の詳細が異なるAPAを検出するために最適化された3’エンドシーケンシングアッセイのクラスがあります。ここで説明するパイプラインはPolyA-seq用に設計されていますが、説明されているように他のプロトコルにも適合させることができます。
本研究では、エクソンベース(DEXSeq9、diffSplice10)とイベントベース(転写産物スプライシングの反復多変量解析(rMATS)11)の2つの大きなカテゴリに分類できる、差動エクソン解析法7,8(図1)のパイプラインを提示します。エクソンベースの方法は、個々のエクソンの条件にわたるフォールド変化を、発現差のあるエクソン使用量を呼び出すための全体的な遺伝子フォールド変化の測定値と比較し、そこからAS活性の遺伝子レベルの測定値を計算します。イベントベースの方法は、エクソン-イントロン-スパニング接合リードを使用して、エクソンスキップやイントロンの保持などの特定のスプライシングイベントを検出および分類し、出力3でこれらのASタイプを区別します。したがって、これらの方法は、AS12,13の完全な分析のための補完的なビューを提供します。DEXSeq(DESeq214 DGEパッケージに基づく)とdiffSplice(Limma10 DGEパッケージに基づく)は、差動スプライシング解析に最も広く使用されているパッケージであるため、研究に選択しました。rMATSは、イベントベースの分析の一般的な方法として選択されました。別の一般的なイベントベースの方法は、MISO(アイソフォームの混合)1です。APAでは、エクソンベースのアプローチを採用しています。
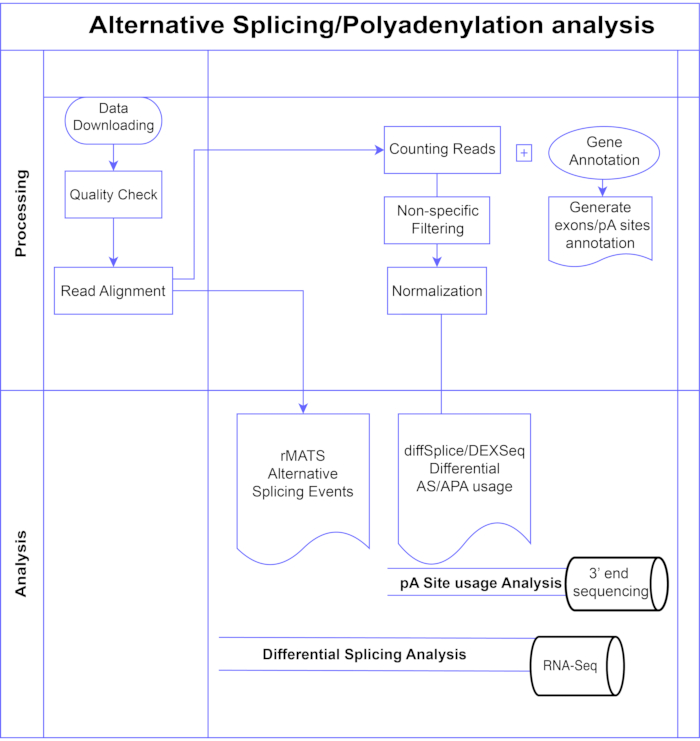
図 1.分析パイプライン。 分析で使用されるステップのフローチャート。手順には、データの取得、品質チェックとリードアライメントの実行、その後の既知のエクソン、イントロン、pAサイトのアノテーションを使用したリードのカウント、低カウントを削除するためのフィルタリング、正規化が含まれます。PolyA-seqデータはdiffSplice/DEXSeq法を用いて代替pA部位について解析し、バルクRNA-SeqはdiffSplice/DEXseq法を用いてエクソンレベルでの選択的スプライシングについて解析し、ASイベントはrMATSを用いて解析した。 この図の拡大版を表示するには、ここをクリックしてください。
本調査で用いたRNA-seqデータは、Gene Expression Omnibus(GEO)(GSE138691)15から取得したものである。この研究のマウスRNA-seqデータを、野生型(WT)とマッスルブラインド様タイプ1ノックアウト(Mbnl1 KO)の2つの条件群で使用し、それぞれ3回の反復を行いました。差的ポリアデニル化部位利用分析を実証するために、マウス胚線維芽細胞(MEF)PolyA-seqデータ(GEOアクセッションGSE60487)16を得た。データには、野生型(WT)、マッスルブラインド様タイプ1/タイプ2ダブルノックアウト(Mbnl1/2 DKO)、Mbnl3ノックダウン(KD)を備えたMbnl 1/2 DKO、およびMbnl3コントロール(Ctrl)を備えたMbnl1/2DKOの4つの条件グループがあります。各条件グループは、2 つの反復で構成されます。
| ゲオアクセッション | SRA 実行番号 | サンプル名 | 条件 | レプリケート | 組織 | シークエンシング | 読み取り長 | |
| RNA-シークエンス | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1ノックアウト | 担当者 1 | 胸腺 | ペアエンド | 100 bp |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1ノックアウト | 担当者 2 | 胸腺 | ペアエンド | 100 bp | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1ノックアウト | 担当者 3 | 胸腺 | ペアエンド | 100 bp | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | 野生型 | 担当者 1 | 胸腺 | ペアエンド | 100 bp | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | 野生型 | 担当者 2 | 胸腺 | ペアエンド | 100 bp | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | 野生型 | 担当者 3 | 胸腺 | ペアエンド | 100 bp | |
| 3P-シーケンセック | GSM1480973 | SRR1553129 | WT_1 | 野生型(WT) | 担当者 1 | マウス胚性線維芽細胞(MEF) | シングルエンド | 40 bp |
| GSM1480974 | SRR1553130 | WT_2 | 野生型(WT) | 担当者 2 | マウス胚性線維芽細胞(MEF) | シングルエンド | 40 bp | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 ダブルノックアウト (DKO) | 担当者 1 | マウス胚性線維芽細胞(MEF) | シングルエンド | 40 bp | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 ダブルノックアウト (DKO) | 担当者 2 | マウス胚性線維芽細胞(MEF) | シングルエンド | 40 bp | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 ダブルノックアウト Mbnl 3 siRNA (KD) | 担当者 1 | マウス胚性線維芽細胞(MEF) | シングルエンド | 40 bp | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 ダブルノックアウト Mbnl 3 siRNA (KD) | 担当者 2 | マウス胚性線維芽細胞(MEF) | シングルエンド | 36 bp | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 ダブルノックアウトとノンターゲティング siRNA (Ctrl) | 担当者 1 | マウス胚性線維芽細胞(MEF) | シングルエンド | 40 bp | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 ダブルノックアウトとノンターゲティング siRNA (Ctrl) | 担当者 2 | マウス胚性線維芽細胞(MEF) | シングルエンド | 40 bp |
表 1.解析に使用したRNA-SeqおよびPolyA-Seqデータセットの概要。
