The analysis of ECs from mesenteric artery using a combination of mechanical and enzymatical dissociation or cryopreservation for use in various downstream assays is depicted here (Figure 1). ECs can be profiled in mesenteric arteries using the following steps: A) mechanical dissociation from the intima coupled with collagenase digestion to culture cells; B) generation of single-cell suspension for scRNA-seq; or C) cross-sections of the artery can be embedded in OCT to be cryosectioned to profile spatial transcriptome (Figure 1 and Figure 2).
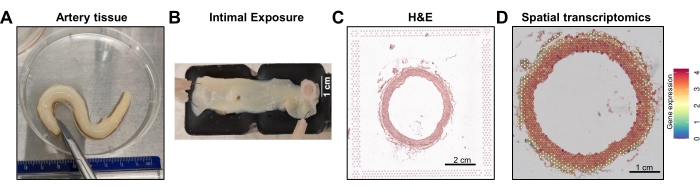
Figure 1: Arterial tissue processing and spatial profiling. (A) A cleaned-up mesenteric artery. (B) Vessel cut open to expose intima. (C) Hematoxylin and Eosin (H&E) staining of mesenteric artery cross-section in the fiducial frame. Image captured using a 5x lens under widefield fluorescence inverted microscope. (D) Representative Space Ranger output file of total gene expression. Please click here to view a larger version of this figure.
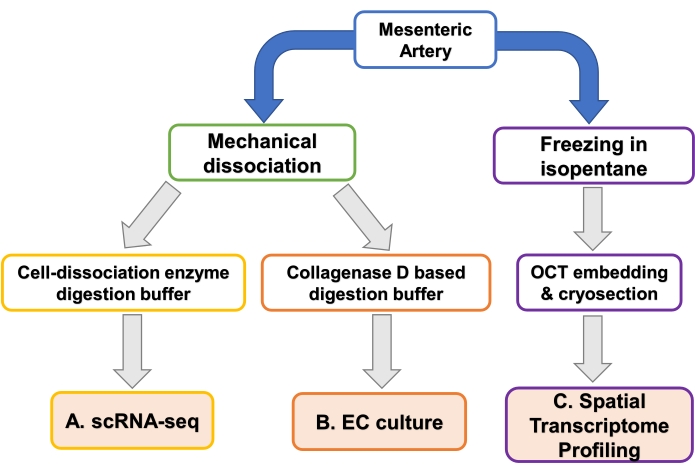
Figure 2: Overview of EC isolation and profiling techniques. Flow diagram showing different methods of processing mesenteric artery. Processing techniques result in cell suspensions suitable for single-cell (sc) RNA-seq, endothelial cell (EC) culture, or whole tissue is embedded in optimal cutting temperature compound (OCT) for spatial transcriptome profiling. Please click here to view a larger version of this figure.
Isolated ECs cultured using the described protocol display a distinct cobblestone-like morphology with minimal contaminating cells (Figure 3A). Expression of EC marker vascular endothelial (VE)-Cadherin was confirmed using immunofluorescence visualizing cell-cell junctions (Figure 3B). Isolated ECs were subjected to flow cytometry analysis for CD31. As negative controls, unstained HUVECs (without antibody) produced a 1.1% signal and HUVECs incubated with IgG yielded a 0.8% signal. As a positive control, HUVECs stained with CD31 antibody displayed 99% purity and were used to gate the channels for human mesenteric arterial endothelial cells (HMAECs). Approximately 80% of cells were CD31 positive indicating HMAECs made up the majority of the freshly isolated cell population (Figure 3C). Unsuccessful isolations, through either scraping too hard/deep, seeding the cells at too low a density, or maintaining the culture beyond passage 3 results in cells with disturbed morphology, potentially expressing mesenchymal markers (αSMA) or elongating resembling a fibroblastic state (Figure 3D).
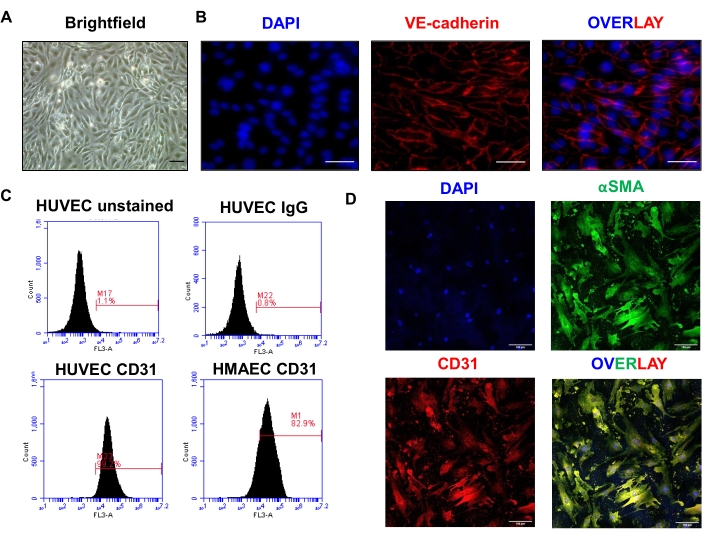
Figure 3: Validation of isolated cultured mesenteric arterial ECs. (A) Brightfield image of isolated ECs, 10x lens, scale bar = 50 µm. (B) Immunofluorescence of VE-Cadherin expression with 4′,6-diamidino-2-phenylindole (DAPI) as a nuclear marker. Image captured using a 10x lens under widefield fluorescence inverted microscope. Representative image shows VE-Cadherin localization. Scale bar = 50 µm. (C) FACS plots of unstained HUVEC (top left), HUVEC stained with IgG control (top right), HUVEC stained with CD31 (bottom left), and the isolated human mesenteric arterial endothelial cells (HMAEC) stained with CD31 (bottom right) (D) Immunofluorescence images of HMAECs with DAPI (nuclear marker), alpha-smooth muscle actin (αSMA) and CD31. 40x lens, scale bar = 100 µm. Please click here to view a larger version of this figure.
ECs isolated using cell-dissociation enzyme underwent scRNA-seq. Figure 4A shows a representative uniform manifold approximation and projection (UMAP) isolated from mesenteric artery using the present protocol for scRNA-seq on isolated HMAECs, with 34% cells clustered as ECs using PECAM1 and VWF (Figure 4B,C respectively) as markers.
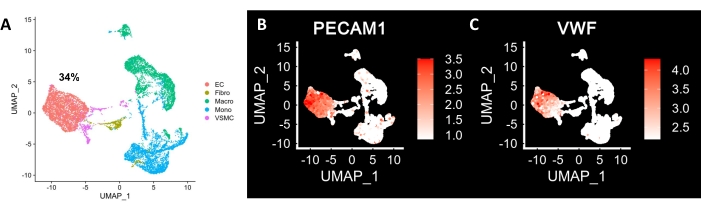
Figure 4: scRNA-seq of mesenteric arterial ECs. (A) Uniform Manifold Approximation and Projection (UMAP) plot of scRNA-seq data from mesenteric artery. A 2-dimensional projection of the manifold in a high dimensional space, where each point represents a single cell and the proximity to other points indicate how similar the transcriptome is with each other. Cells are color-coded by their cell types. (B) PECAM1 expression projected onto UMAP. (C) VWF expression projected onto UMAP. Color bars represent the expression of levels of genes where the RNA levels are depicted by log-normalized unique molecular identifier counts. Please click here to view a larger version of this figure.
For spatial transcriptomic workflow, extracted RNA from tissue sections was electrophorized on a gel to visualize RNA quality. A weak signal or absence of 18S and 28S ribosomal RNA bands, and the presence of degradation products observed in Figure 5A, lane A1, is an example of poor-quality RNA and should not be processed further. Samples with a concentration out of the range could lead to lower RIN (Figure 5A, lane B1). However, diluting the RNA by two times increased RIN sufficient for proceeding (Figure 5A, lane C1). Before determining gene expression, a tissue optimization was performed to determine the optimal permeabilization time to release RNA to be captured by the oligonucleotides on the gene expression slide (Figure 5B). Based on the fluorescence imaging of complementary DNA (cDNA) footprint, 18 min permeabilization produced the lowest background but strongest signal, and thus was selected to be the optimal duration. Hematoxylin and eosin (H&E) staining visualized the morphology of the vessel allowing regions of interest to be identified (Figure 5C). Library quality was assessed (Figure 5D) and determined to be suitable for sequencing. UMI counts per spot, number of reads per spot, and total genes are quality metrics produced by the Space Ranger pipeline, the variability of each displayed in Figure 5E. Finally, gene expression (using Actin alpha-2 (ACTA2) as an example, Figure 5F) was visualized with spatial anchoring on the H&E stained vessel.
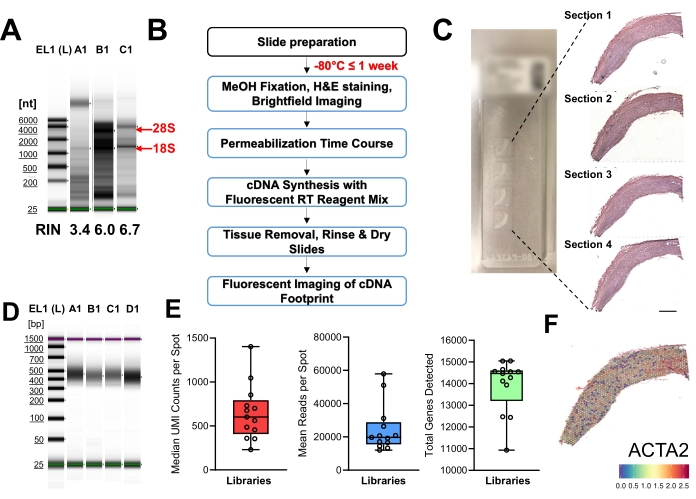
Figure 5: Spatial transcriptomic workflow and representative results (A) Quality assessment of RNA extracted from two samples of mesenteric artery, A1 from one sample, B1 from the second sample, C1 diluted 2x from B1. Red arrows indicate ribosomal 28S and 18S bands. (B) Spatial tissue optimization (TO) workflow. (C) Gene expression (GEX) slides showing the tissue sections (left panel); H&E images of the tissue sections loaded on the GEX slide before permeabilization for 18 min, reverse transcription and library construction (right panel), scale bar = 1 cm. (D) Quality assessment of the libraries prepared. (E) Output metrics from Space Ranger show ranges between average reads, UMIs, and genes per spot for different libraries. Error bars denote min and max. (F) ACTA2 expression across vessel section. All microscopy images were taken using a widefield inverted microscope, from a 5x lens, tilescan module.Please click here to view a larger version of this figure.
