Our understanding of cell differentiation and the genesis of tissues and organs is the result of decades of elaborate targeted screens of genes and their products. Increasing our knowledge of all the biomolecules and their quantities during important cellular events would help unravel molecular mechanisms that control the spatial and temporal patterning of the vertebrate body plan. Technologies enabling molecular amplification and sequencing are now able to routinely report on large numbers of genes and transcripts, supporting hypothesis-driven studies in basic biological and translational research. To understand developing systems, a complex relationship between transcription and translation advocates for direct analysis of multiple proteins and their post-translational modifications. Global proteomics using in vitro biological systems, such as induced pluripotent stem cells, began to delineate mechanisms of tissue induction1,2. In complex organisms, such as the vertebrate embryo, development relies on morphogen gradients in the context of space and time3. It follows that gaining knowledge of proteomic changes as cells differentiate to form specialized tissues, such as neural tissues, offers a key to unlock molecular programs controlling normal and defective development and guide next-generation therapeutics.
The vertebrate South African clawed frog (Xenopus laevis) is a well-established model in cell and developmental, neuro-, and regenerative biology. Sir John Gurdon's 2012 Nobel Prize in Physiology or Medicine4,5 for the discovery of pluripotency of the somatic nucleus highlighted the importance of this model for discoveries in basic and translational studies. Xenopus embryos develop externally to the mother, thus facilitating direct manipulation of cells, cell clones, and gene expression over various stages of development. Asymmetrical pigmentation and stereotypical cell divisions enabled the charting of reproducible fate maps from the 16-6 and 32-cell7,8 stage embryo. For high-resolution mass spectrometry (HRMS) based proteomics, additional advantages of the model include relatively large size (~1 mm in diameter), which yields abundant protein content for analysis (~130 µg in early cleavage-stage embryos, ~10 µg of protein content in single cells of the 16-cell embryo)9,10.
At present, HRMS is the leading technology of choice for detecting proteins. This technology enables direct, sensitive, and specific detection and quantification of multiple, usually hundreds-to-thousands of different proteins11. Bottom-up proteomics by HRMS involves a series of interconnected steps. Following extraction from the cell/tissue sample, proteins are digested with a proteolytic enzyme, such as trypsin (bottom-up proteomics). The resulting peptides are separated based on their different physicochemical properties, including hydrophobicity (reversed-phase liquid chromatography, LC), net charge (ion-exchange chromatography), size (size exclusion chromatography), or electrophoretic mobility (capillary electrophoresis, CE). Peptides are then charged (ionized), typically using electrospray ionization (ESI), and peptide ions are detected and sequenced via gas-phase fragmentation by tandem HRMS. The resulting peptide data are mapped to the proteome of the organism being studied. With protein-specific (proteotypic) peptide ion signal intensity correlating with concentration, protein quantification can be performed label-free or label-based (multiplexing quantitation). HRMS proteomics yields a rich resource of information on the molecular state of the system under study, allowing for the generation of hypotheses and follow-up functional studies.
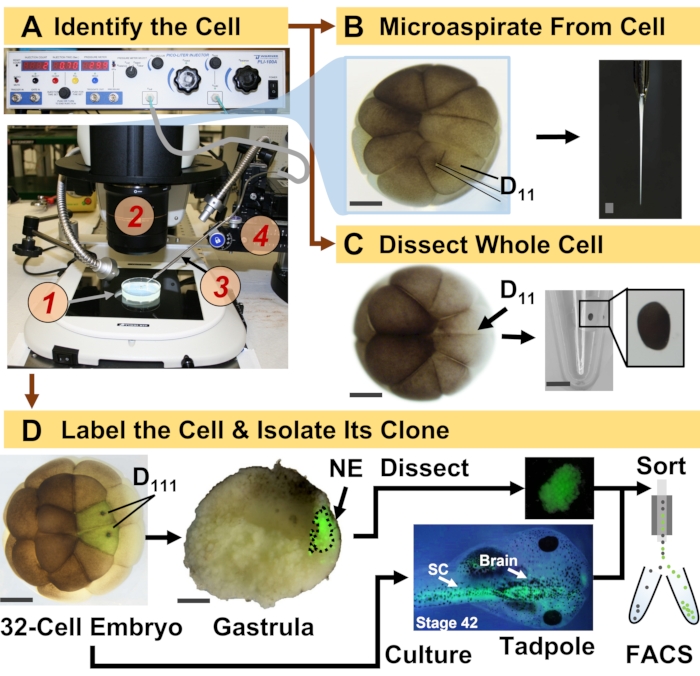
Figure 1: Spatiotemporally scalable proteomics enabling cell-lineage guided HRMS proteomics in the developing (frog) embryo. (A) Visualization of the specimen (1) using a stereomicroscope (2) for injection of an identified cell (inset), using a fabricated micropipette (3) under control by a translation-stage (4). (B) Subcellular sampling of the identified left D11 cell in a 16-cell embryo. (C) Dissection of a whole D11 cell from a 16-cell embryo. (D) Fluorescent (green) tracing of the left and right D111 progenies from a 32-cell embryo to guide dissection of the neural ectoderm (NE) in the gastrula (stage 10) and isolation of the descendent tissue from the tadpole using FACS. Scale bars: 200 µm for embryos, 1.25 mm for the vial. Figures were adapted with permission from references15,19,21,59. Please click here to view a larger version of this figure.
The protocol presented here enables HRMS-based quantification of large numbers of proteins in identified cells/tissues in developing X. laevis embryos. The approach builds on accurate cell identification, reproducible cell fate maps, and established methodologies to track cell lineages in this biological model6,7,8. As shown in Figure 1, we study proteomes from single cells by employing whole-cell dissection or capillary microsampling to aspirate cellular content. Monitoring the lineage of a cell permits us to study the spatiotemporal evolution of the proteome as cells form tissues during gastrulation. The cell progeny is fluorescently marked by injecting a fluorophore conjugated to inert dextran or mRNA for fluorescent protein (e.g., green fluorescent protein, or GFP). The labeled progeny is isolated at desired developmental time points. During gastrulation, cell clones that are tightly clustered may be isolated by dissection. After gastrulation, cell clones may be distributed within the embryo owing to migratory movements and can be isolated from dissociated tissues by fluorescence-activated cell sorting (FACS). Proteins in these cells and tissues are measured via bottom-up proteomics employing HPLC or CE for separation and ESI tandem HRMS for identification. Cell-lineage-guided HRMS proteomics is scalable to different cell sizes and lineages within the embryo and is specific, sensitive, and quantitative. Through select examples shown here, we also demonstrate this protocol to be scalable and broadly adaptable to different types of cells and cell lineages.
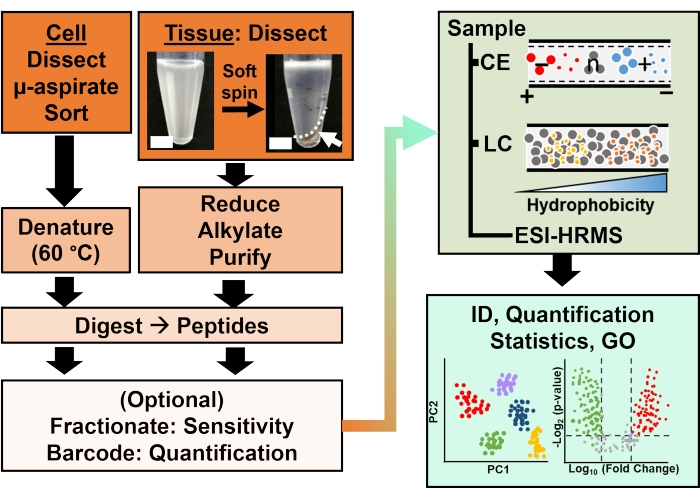
Figure 2: The bioanalytical workflow. Micro-dissection and capillary aspiration, or FACS facilitated sampling of cellular and clonal protein content. Depletion of abundant yolk proteins and separation by capillary electrophoresis (CE) or nano-flow liquid chromatography (LC) enhanced identification (ID) sensitivity using electrospray ionization (ESI) high-resolution mass spectrometry (HRMS). Quantification revealed dysregulation, supplying new information for hypothesis-driven studies in conjunction with information available from gene ontology (GO). Figures were adapted with permission from reference15. Please click here to view a larger version of this figure.
This protocol enabled the study of proteins in single cells and their lineages as they establish tissues in X. laevis embryos. Figure 1 illustrates one such application of the approach to study proteins in neural-tissue-fated cells and the newly induced neural ectoderm in the embryo. As shown in Figure 1A, the bioanalytical workflow integrated traditional tools of cell and developmental biology to identify, inject/aspirate cells, and collect specimens. Figure 1B shows microprobe sampling of the left dorsal-animal (D11) cell in the 16-cell embryo in vivo using a microinjector; after the experiment, embryos successfully developed to tadpoles with normal anatomy56. Large embryonic cells (~100-250 µm in diameter) were conducive to manual microdissection as well. Dissection of a right dorsal-animal midline (D11) cell is illustrated from the 16-cell embryo in Figure 1C. The setup also permitted tracing a clonal trajectory by injecting a fluorescent tracer into the identified precursor. As shown in Figure 1D, the approach allowed to isolate clones arising from the left and right D111 cells via tissue dissection or by fluorescence-activated cell sorting (FACS). The sample collection strategies described here are sufficiently scalable in space and time to study embryonic development in new details.
The bioanalytical workflow integrated HRMS technologies to improve sensitivity and quantification (Figure 2). The collected proteins were measured via a bottom-up proteomic approach. Optional fractionation of samples based on orthogonal chemistry (e.g., high-pH then low-pH reversed-phase LC) aided detection sensitivity. To separate peptides, CE was selected for trace amounts of samples (<<~100 ng)) and nanoLC for limited amounts of materials (>>150 ng). Peptides were sequenced using ESI-HRMS. The detected proteins were quantified using label-free and label-based strategies. Simplification of sample processing for single-cell analysis, such as elimination of the typical steps of reduction and alkylation followed by CE analysis, facilitated the identification of ~400-800 different proteins. Among the reported proteins were many with important functional roles, such as chaperonin containing TCP1 subunit 3 (Cct3), voltage-dependent ion channel (Vdac2), and creatine kinase-brain (Ckb). Multivariate and statistical data models helped us10,57 and others9,58 find previously unknown differences in the proteomic composition of select cells and tissues using the approaches summarized in this protocol. Notably, these HRMS measurements required no functioning probes or knowledge about the composition of the specimens ahead of time, supporting discoveries.
In a series of studies, the proteomic state of identified cells in developing embryos of X. laevis (Fig. 3) was quantified21. Figure 3A shows the detection of gene translational differences between D11 cells that were dissected from different embryos10. Microsampling CE-ESI-HRMS allowed us to identify and quantify up to ~700 proteins from single cells21. The representative primary HRMS-MS/MS data on identifying ~400 cumulative proteins from technical duplicate measurements on a D11 cell in the embryo was deposited to PRIDE. The approach was scalable to smaller cells and embryos from other model organisms, such as zebrafish21. Scalability to smaller cell sizes allowed us to explore the spatiotemporal reorganization of the D11 progeny from a live embryo as it developed in time. Figure 3B presents the use of this protocol to perform subcellular quantitative proteomics of identified cells in the 16-, 32-, 64-, and 128-cell embryos. Proteins were separated into four groups that displayed distinct abundance profiles over clonal development21.
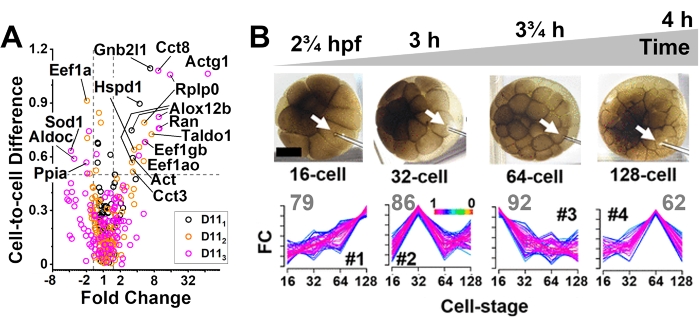
Figure 3: Protocol scalability from the subcellular space to clonal tissues in X. laevis embryo. (A) Measurement of proteomic differences between identified whole D11 cells, revealing cell-cell heterogeneity. Gene names shown for select proteins. (B) Reorganization of the cellular proteome in the developing D11 cell clone. Fuzzy C-means cluster analysis (GProx) of protein dynamics, grouping proteins based on similar expression patterns. Gray numbers show the number of different proteins that were quantified in each cluster. Figures were adapted with permission from references21,57. Please click here to view a larger version of this figure.
This protocol allowed us to conduct spatial and temporal proteomics in identified, developing cell clones (Figure 4). Figure 4A demonstrates the application of the approach for labeling and isolating cell clones constituting two tissues with important roles in neural tissue development and patterning of the embryo. The majority of the Spemann organizer (SO) was traced by labeling the left and right D112 and D212 cells via injection of fluorescent dextran. In parallel, the neighboring dorsal-animal (D111) cells were labeled to mark the majority of the neural ectoderm (NE). The tissues were dissected, and their protein content was analyzed following this protocol. nanoLC-ESI-HRMS returned up to 2,000 different proteins from the tissues, including signaling, such as the Wnt, Fgf, and Tgfβ pathways. The representative primary HRMS-MS/MS data on identifying ~1,800 cumulative proteins from technical duplicate measurements on a pool of dissected SO tissues was deposited to PRIDE. The Wnt pathway ligand Wnt10a and Wntless (Wls), a membrane protein dedicated to the secretion of Wnt ligands, were detected only in the NE proteome. The Wnt pathway was found suppressed in the SO and may explain a lack of Wnt-interacting proteins detected in the SO. These results showcase the applicability of this protocol for studying lineage-specific differences within the embryo.
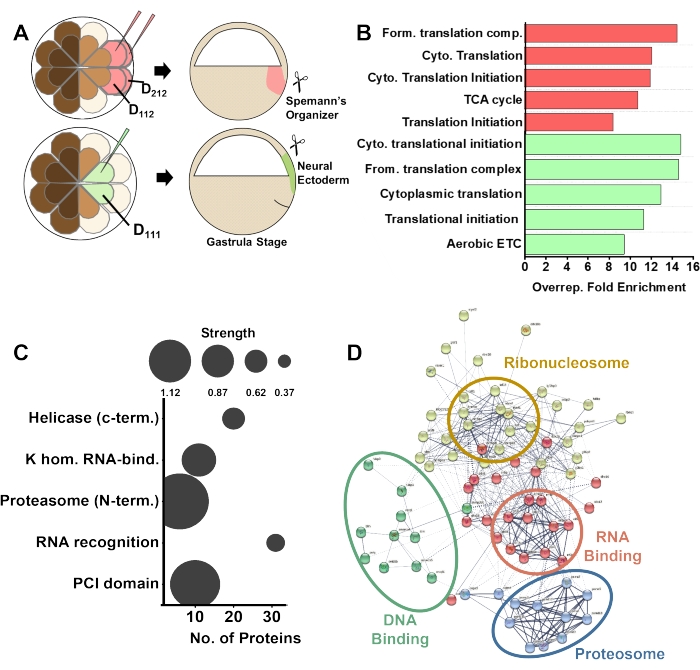
Figure 4: Example of data analysis from spatial tissue proteomics in the X. laevis embryos. (A) Differential labeling of the Spemann's Organizer (SO) and the neural ectoderm (NE) tissues by injection of fluorescent protein mRNA in the predecessor D112 and D212 in the 32-cell embryo, respectively. (B) Top 5 overrepresented biological processes in the SO (red) and NE (green) proteome showing detectable differences. Pathway overrepresentation analysis shows biological processes using Bonferroni correction. (C) Protein domain enrichment analysis (SMART), revealing enrichment of DNA and RNA binding motif-containing proteins in the SO. (D) STRING analysis predicting canonical protein-protein interactions based on the detected SO proteome. Please click here to view a larger version of this figure.
The diverse proteomic data serve as valuable information for the assessment of function. The proteomic data can be evaluated using canonical knowledge bases. As shown in Figure 4B, pathway analysis of dysregulated proteins showed overrepresented translation and energy metabolism in both the SO and the NE datasets in our recent studies. The NE proteome was enriched in proteins associated with nuclear transport of protein cargo in the cell, likely indicating downstream events following signaling in the newly established NE (Figure 4C). The enrichment analysis in Figure 4D found upregulation in translation initiation, RNA-binding, binding in the proteasome complex, suggesting a role for dynamic protein turnover developing SO. Cell lineage-guided HRMS proteomics is scalable in space and time and sufficiently sensitive to help better understand the molecular organization of cells during normal and impaired development.
Supplementary File. Please click here to download this File.
