Single-Cell Proteomics Preparation for Mass Spectrometry Analysis Using Freeze-Heat Lysis and an Isobaric Carrier
Summary
In this protocol, we describe how to prepare mammalian cells for single-cell proteomics analysis via mass spectrometry using commercially available reagents and equipment, with options for both manual and automatic pipetting.
Abstract
Single-cell proteomics analysis requires sensitive, quantitatively accurate, widely accessible, and robust methods. To meet these requirements, the Single-Cell ProtEomics (SCoPE2) protocol was developed as a second-generation method for quantifying hundreds to thousands of proteins from limited samples, down to the level of a single cell. Experiments using this method have achieved quantifying over 3,000 proteins across 1,500 single mammalian cells (500-1,000 proteins per cell) in 10 days of mass spectrometer instrument time. SCoPE2 leverages a freeze-heat cycle for cell lysis, obviating the need for clean-up of single cells and consequently reducing sample losses, while expediting sample preparation and simplifying its automation. Additionally, the method uses an isobaric carrier, which aids protein identification and reduces sample losses.
This video protocol provides detailed guidance to enable the adoption of automated single-cell protein analysis using only equipment and reagents that are widely accessible. We demonstrate critical steps in the procedure of preparing single cells for proteomic analysis, from harvesting up to injection to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Additionally, viewers are guided through the principles of experimental design with the isobaric carrier, quality control for both isobaric carrier and single-cell preparations, and representative results with a discussion of limitations of the approach.
Introduction
Single-cell analysis is widely used to study the level of heterogeneity within biological systems that would otherwise be indiscernible by bulk measurements1,2,3. Such cellular diversity can have functional consequences that can further the understanding of integral biological processes, from cancer cell therapeutic resistance4,5,6 to cell-to-cell heterogeneity in diabetes7,8,9,10. Many investigations have focused on measuring nucleic acids in single cells, by measuring genetic or transcriptomic levels and have enabled the classification of cell types and states. Such progress in the nucleic acid space, however, cannot fill the knowledge gap of post-transcriptional regulation11, driving the need for similarly high-throughput protein measurements in single cells12,13,14,15.
We have developed SCoPE216,17 to address the demand for rigorous and robust single-cell proteomics of mammalian systems. It is a multiplexed method, allowing for increased throughput by labeling and analyzing many cells in parallel18, in contrast to label-free mthods that analyze one cell at at time19,20. The sample preparation methodology, mass spectrometry approach, and subsequent data analysis steps allow for accurate quantification of hundreds to thousands of proteins per single cell. This method makes mass spectrometry analysis of single cells possible through the isobaric carrier21, a small bulk sample of cells (usually 25-200) that is biologically similar to the single-cell population of interest. This carrier material is multiplexed in a set with a reference channel of the same material and single cells through the use of tandem mass tags (TMT labels). The carrier channel reduces sample loss to surface areas and provides the ion backbone fragments for successful peptide identification. The reference channel, which is prepared in bulk and subsequently diluted down to 5-10 cell equivalents for each single-cell set, helps control technical variability in the analysis. Specifically, the reference allows for the normalization of variability caused by LC-MS/MS-related effects, such as ion sampling and ionization efficiencies. Usually, the reference and the carrier channels are made from the same cell population.
This sample preparation protocol is a second-generation method building upon SCoPE-MS22 by multiple synergistic improvements. The improvements include a cell lysis method that avoids sample clean-up, which is a common step of MS proteomics preparations. It uses mPOP (minimal ProteOmic sample Preparation)23, which is a freeze-heat lysis method. This method enabled lysis of single cells in smaller volumes and in multiwell plates rather than in vials, facilitating a higher throughput rate and automation by thermal cyclers. Altogether, this method has lowered the cost per single cell and increased quantitative accuracy as compared to SCoPE-MS16,24.
This protocol describes how to prepare single cells for proteomic analysis, including how to prepare the carrier and reference channels using TMTpro 18-plex isobaric labels. Mammalian cells that are in single-cell suspension and that can be isolated into 384-well plates are likely amenable to this protocol. Also included are representative results of successful single-cell sets, displaying several quality control plots generated by a R Shiny app, DO-MS25. Other published software tools26,27 and experimental guidelines17,21 have improved the adoption of this protocol. We hope this visual guide will further help researchers perform single-cell proteomics experiments.
Protocol
NOTE: In this protocol, room temperature (RT) is 18-22 °C.
1. Carrier and reference material generation
- Cell isolation
- Collect cells of interest into cell suspensions in ice-cold 1x PBS.
NOTE: If possible, the carrier and reference material should be prepared from the cell population(s) chosen to be studied at the single-cell level. Similar numbers of cells from the different experimental conditions (such as stimulated and unstimulated immune cells) should constitute the carrier and reference. In situations in which a large enough number of these cells cannot be harvested, a suitable cell population should be selected based on biological similarity. - Using a hemocytometer or other means of cell counting (such as FACS), count the number of cells in the suspension.
- Spin down and resuspend 22,000 cells of each cell type in 11 µL of HPLC-grade water, separately into PCR tubes (standard 250 µL PCR tubes).
NOTE: The number of cells advised here are sufficient for the carriers and references for the number of sets that can be prepared from a 384-well plate full of single cells. The cell input in this step and reagent amounts in subsequent steps can be scaled proportionally for greater numbers of sets. The speed of spinning down is dependent on the cell culture practices of the lab. A typical spin down: 300 × g for 5 min. - Transfer the samples to a -80 °C freezer for at least 30 min.
PAUSE POINT: Samples can stay frozen for several months with no consequences for single-cell data.
- Collect cells of interest into cell suspensions in ice-cold 1x PBS.
- Cell lysis
- Heat the PCR tube with the cells to 90 °C for 10 min (with thermal cycler lid set at 105 °C), and then let the tubes cool to 12 °C right after the heating cycle.
- Vortex the tube briefly, and then spin down in a bench-top PCR tube spinner at RT to collect all the liquid at the bottom of the tube.
- In a water bath sonicator, sonicate the tubes for 5 min, then place them on ice at RT.
- Trypsin digestion
- Add 2.2 µL of the master mix (see Table 1 for the components) to each sample tube while on ice.
CAUTION: Trypsin can cause skin, respiratory, and eye irritation. Be sure to wear personal protective equipment. Handle under a chemical fume hood. - Vortex the tubes briefly to mix and spin down in a bench-top PCR tube spinner at RT to collect all liquid at the bottom of each tube.
- Heat the samples at 37 °C for 3 h (with the thermal cycler lid set at 52 °C).
- Add 2.2 µL of the master mix (see Table 1 for the components) to each sample tube while on ice.
- TMT labeling reaction
- Spin down the samples after digestion in a bench-top PCR tube spinner at RT.
- Split each sample into two equal volumes of 6.6 µL each, so that each tube contains 11,000 cells.
- To the tubes meant for the carrier, add 3.3 µL of TMT label 126 that has been resuspended at a concentration of 85 mM.
- To the tubes meant for the reference, add 3.3 µL of TMT label 127N that has been resuspended at a concentration of 85 mM.
- Vortex the tubes briefly and spin down in a bench-top PCR tube spinner at RT to collect the liquid at the bottom.
- Leave the tubes at RT for 1 h.
- Quenching of TMT labeling reaction
- Add 1.65 µL of 0.5% hydroxylamine to each tube.
CAUTION: Hydroxylamine can cause skin irritation. Be sure to wear personal protective equipment. - Vortex the tubes briefly and spin down in a bench-top PCR tube spinner at RT to collect the liquid at the bottom.
- Leave the tubes at RT for 30 min.
- Add 1.65 µL of 0.5% hydroxylamine to each tube.
- Combination of carrier and reference
- If samples that will constitute the carrier have been separately labeled, mix them in equal ratios.
- If samples that will constitute the reference have been separately labeled, mix them in equal ratios.
- Mix the carrier and reference so that the final concentration of the carrier is 100-200 cells/µL, and the reference concentration is 5-10 cells/µL.
- Assess the quality of the carrier and reference materials prior to combining them with single-cell sets.
NOTE: Quality control of the carrier and reference material can be carried out by a variety of methods, but it is recommended to use the DO-MS software, available at do-ms.slavovlab.net17. Critical measurements of quality include miscleavage rate (<25%, a metric calculated as the number of confidently identified peptides with a missed cleavage divided by the number of confidently identified peptides in total) and labeling efficiency (>99%, a metric calculated as the number of labeling sites, namely the peptide N-terminus and Lysine, with a label divided by the total number of labeling sites).
PAUSE POINT: The carrier and reference materials can be stored at -80 °C until required.
| Component | Mix Concentration | Final concentration per tube |
| Trypsin Gold | 50 ng/µL | 8.33 ng/µL |
| TEAB, pH = 8.5 | 500 mM | 83.33 mM |
| Benzonase nuclease | 1.2 units | 0.2 units |
Table 1: Reagent amount of master mix. The final concentrations of reagents needed for trypsin digestion are listed.
2. SCoPE2 sample preparation
- Cell isolation
- Supplement HPLC-grade water with 25 fmol per peptide per µL of a synthetic peptide solution.
NOTE: Waters MassPrep peptide solution is an example of what can be used. It is composed of seven peptides that do not ionize very well. This is a key aspect of this solution: these peptides do not interfere with accurate mass spectrometry quantitation of single cells. Any highly purified mixture of a few peptides unlikely to be in the samples of interest will be appropriate. - Add 1 µL of this mix to each well of a 384-well plate using a liquid-dispensing robot or manual pipette.
- Seal the plate and spin it down in a bench-top plate spinner at RT to collect the liquid at the bottom.
PAUSE POINT: Plates with this solution can be stored at -80 °C until required. - Obtain a suspension of unfixed cells that are disaggregated.
NOTE: How exactly the cell suspension is obtained is based on the cell type of interest. Here are broad examples:- Suspension cells: spin down the cells into a pellet, and remove the cell culture medium.
- Adherent cells: remove the cells from the dish or flask through trypsinization or scraping. Then, spin them down to remove the cell culture medium.
- Wash the cell suspension twice with 1x ice-cold PBS. Leave the cells suspended in 1x PBS after the second wash.
- If the 384-well plate was frozen, thaw it out at RT. Spin it down to ensure all liquid is at the bottom of the wells.
- Use a cell sorter or other available means, such as manual handpicking, to distribute single cells into respective wells.
NOTE: Add single cells to wells while leaving some wells empty for negative and positive controls (see discussion). When using flow cytometry, use the lowest possible flow rate for the flow cytometer. Lower flow rate is thought to be gentler for cell handling. Additionally, the nozzle size should be appropriate for use with the cells of interest. It is recommended to collaborate with a flow cytometry facility. - Add positive controls of 2-5 cell equivalents lysate to selected wells pipetted manually or with liquid handling robots.
- Seal and spin down the plate with sorted cells in a bench-top plate spinner at RT to collect the liquid at the bottom. Freeze the plate at -80 °C as soon as possible.
PAUSE POINT: Plates with sorted single cells can be stored at -80 °C until required.
- Supplement HPLC-grade water with 25 fmol per peptide per µL of a synthetic peptide solution.
- Cell lysis
- Take the 384-well plate with the sorted single cells and put it into the thermal cycler as quickly as possible.
- Heat the plate to 90 °C for 10 min (with the lid temperature set to 105 °C), and then let the plate cool to 12 °C.
- Spin down the plate briefly in a bench-top plate spinner at RT.
- In a water bath sonicator, sonicate the plate for 5 min at RT, then place it on ice.
- Trypsin digestion
- Prepare 100 µL of the master mix (see Table 1 for components) per 384-well plate.
- To each well of the 384-well plate, add 0.2 µL of the master mix prepared in step 2.3.1 using a liquid handler.
NOTE: Use larger volumes if manual pipetting is used, making sure that the final concentrations of reagents are still the same per well. - Seal the plate, vortex for 5 s, and spin down in a bench-top plate spinner at RT.
- Heat the 384-well plate at 37 °C (with the lid temperature set to 52 °C) for 3 h.
- TMT labeling reaction
- Take the 85 mM stocks of TMT labels (128N through 135N) out of the -80 °C freezer. Warm the tubes to room temperature before opening them.
- Dilute the labels to 22 mM in anhydrous acetonitrile.
CAUTION: Acetonitrile is a flammable liquid. It can irritate the skin, eyes, respiratory tract, and central nervous system. Be sure to wear personal protective equipment. Handle under a chemical fume hood. - Using this labeling strategy, add 0.5 µL of diluted TMT labels to each well of the 384-well plate. Use either a liquid-dispensing robot (if using a liquid handler, be sure to use associated equipment that is suitable for organic solvents) or a manual pipette.
NOTE: See Table 2 for the labeling strategy. See Table 3 for an example plate layout. - Seal the plate, vortex for 5 s, and spin down in a bench-top plate spinner at RT.
- Let the labeling reaction proceed at RT for 1 h.
- Quenching of TMT labeling reaction
- To each well of the 384-well plate, add 0.2 µL of 0.5% hydroxylamine (diluted in HPLC-grade water) using a liquid handler.
NOTE: Use larger volumes if manual pipetting is used, making sure that the final concentrations of reagents are still the same per well. - Seal the plate, vortex for 5 s, and spin down in a bench-top plate spinner at RT.
- Keep the plate at RT for 30 min.
- To each well of the 384-well plate, add 0.2 µL of 0.5% hydroxylamine (diluted in HPLC-grade water) using a liquid handler.
- Preparation for LC-MS/MS analysis: combination of single cells and control wells with carrier/reference material
- Remove the combined carrier/reference material from the -80 °C freezer.
- For each set, pipette 1 µL of carrier and reference material into the first well that will be part of a single set. Pipette the complete volume of the first well to the subsequent well. Continue pipetting the complete volume from one cell to the next until all wells to be included in a single set have been combined in the final well.
NOTE: The carrier and reference material should be at a concentration of 200- and 5-cell equivalents, respectively, but as discussed previously21, these amounts may vary. Each set, as outlined in the labeling strategy (see Table 2), should contain carrier, reference, and up to 12 or 14 single-cell or control samples when using TMTpro-16plex or TMTpro-18plex, respectively (see step 2.4.3). - For each set, add 5 µL of 50% acetonitrile (diluted in HPLC-grade water) to the first single-cell well to be included in each set. Pipet the complete volume from the first well to the subsequent well. Continue pipetting the complete volume from one cell to the next until all wells to be included in a single set have been combined in the final well.
NOTE: This washing step can help in sample recovery from the wells. - Transfer each set into their individual autosampler glass inserts.
NOTE: These sets can also be combined into single wells of a 384-well plate and directly placed in the autosampler for injection28. - Once all sets are combined and placed into glass inserts, dry the samples.
PAUSE POINT: Dried sets can be stored at -80 °C for at least 1 week prior to running on a mass spectrometer. - Prior to LC-MS/MS analysis, add 1.2 µL of 0.1% formic acid (diluted in HPLC-grade water) to each set to resuspend the labeled peptides.
CAUTION: Formic acid is a flammable liquid. It can cause serious eye damage or skin burns. Be sure to wear personal protective equipment. Handle in a well-ventilated area. - Place the autosampler inserts into glass autosampler vials and close each sample with a cap.
- Vortex each vial for 5 s to make sure resuspension is complete, and then spin down in a vial spinner at RT.
- Ensure that each sample is at the bottom of the insert, rather than splashed on the sides.
- Place the vials in the autosampler tray.
- For each set, inject 1 µL for LC-MS/MS analysis.
| Label | 126 | 127N | 127C | 128N | 128C | 129N | 129C | 130N | 130C | …. | 135N |
| Sample Type | Carrier | Reference | Empty | Empty | SC/Control | SC/Control | SC/Control | SC/Control | SC/Control | …. | SC/Control |
Table 2: Example SCoPE2 TMTpro-18plex labeling scheme. The carrier and reference are usually labeled in the first two TMT labels. Then, the next two labels are skipped due to potential isotopic contamination that would make the quantitation less accurate. The other labels left are for single cells or controls.
| 128C | 129N | 129C | 130N | 130C | 131N | 131C | 132N | 132C | 133N | 133C | 134N | 128C | 129N | 129C | 130N | 130C | 131N | 131C | 132N | 132C | 133N | 133C | 134N | |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | |
| A | SC | SC | – | SC | SC | SC | SC | SC | + | SC | SC | SC | SC | SC | SC | SC | SC | SC | – | SC | + | SC | SC | SC |
| B | SC | + | SC | SC | SC | SC | SC | SC | SC | SC | + | SC | SC | SC | SC | SC | SC | SC | SC | SC | – | SC | SC | + |
| C | SC | – | SC | – | SC | SC | SC | + | SC | – | SC | SC | SC | SC | + | SC | – | SC | SC | SC | SC | SC | SC | – |
| D | SC | SC | SC | SC | SC | SC | – | SC | SC | SC | SC | + | SC | – | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC |
| E | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | + | SC | SC | SC | SC | SC | – | SC | SC | |
| F | SC | SC | SC | SC | SC | SC | SC | SC | – | SC | SC | SC | SC | + | SC | SC | SC | + | SC | SC | SC | + | SC | SC |
| G | SC | SC | SC | + | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | – | SC | SC | SC | SC |
| H | SC | SC | SC | SC | SC | + | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | + | SC | SC | SC | SC | SC | SC | SC |
| I | SC | SC | SC | SC | SC | SC | SC | SC | SC | + | SC | SC | – | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC |
| J | – | SC | + | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | – | SC | SC | SC | SC | SC | SC |
| K | SC | SC | SC | SC | – | SC | SC | SC | SC | – | SC | SC | + | SC | – | – | SC | SC | SC | SC | SC | SC | SC | SC |
| L | SC | SC | SC | SC | SC | – | SC | SC | SC | SC | SC | – | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | + | SC |
| M | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | + | SC | SC | SC | SC |
| N | + | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC |
| O | SC | SC | SC | SC | + | SC | SC | SC | SC | SC | – | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC |
| P | SC | SC | – | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | SC | + | SC | SC | SC | – | SC |
Table 3: Example plate layout for labeling using TMTpro-16plex. Using a 384-well plate format (or any other plate format), it is important to randomize the wells in which cells are sorted into. A different plate layout will be used for TMTpro-18plex.
Representative Results
Positive results from the protocol entail verifying that a number of critical steps in the protocol have been successfully performed; these include the preparation of the carrier, single-cell isolation, lysis, digestion, barcode labeling, and MS parameter optimization. DO-MS plots allow the easy assessment of the completion of each critical step in the protocol. A successfully prepared carrier, which usually corresponds to 25 and 200 cells in quantity (per set), is well-digested and well-labeled. Plots from DO-MS allow quantifying the intensity of the sample (to estimate the quantity), the digestion efficiency (ideally <25% miscleavages), and the labeling efficiency (must be >99%). Most critically, carrier samples that are not completely labeled may permit the cross reaction with the barcodes intended for single cells and add a spurious signal to the quantification of single-cell peptides.
Negative control wells include all experimental reagents but lack a cell. This allows not only background noise to be assessed but also cell isolation efficiency to be determined by providing a representative signal of an empty well. The DO-MS report offers plots to assess the measured signal of the control well alongside the single-cell wells to determine the success of cell isolation and background noise. Additionally, the quality of quantitation can be assessed by using the SCoPE2 pipeline (available on GitHub: github.com/SlavovLab/SCoPE2) or the SCP Bioconductor package29.
Digestion and labeling efficiency of the single cells may not be directly assayed as with the carrier, but DO-MS again provides plots that allow the estimation of the digestion and labeling efficiency. By including a control of 100-200 cells alongside the preparation of the single cells (i.e., receiving all the same reagent additions), the digestion and labeling efficiencies can be assessed for that control, and assumed to be shared by the single cells prepared alongside. Poor digestion will be indicated by a high ratio of the intensity of miscleaved peptides to their completely cleaved counterparts (Figure 1).
Another factor to consider in the sets is the fraction of missing data and the median reporter ion intensity in each TMT channel (Figure 2). When single cells are successfully prepared, the amount of missing data per cell and positive control is much lower than in negative control samples. Likewise, the median intensity of precursors is much higher in single cells and positive controls than for negative controls. The optimization of MS parameters has been discussed extensively elsewhere21, and optimal parameters can be determined using such tools as DO-MS or SCP Companion25,27.
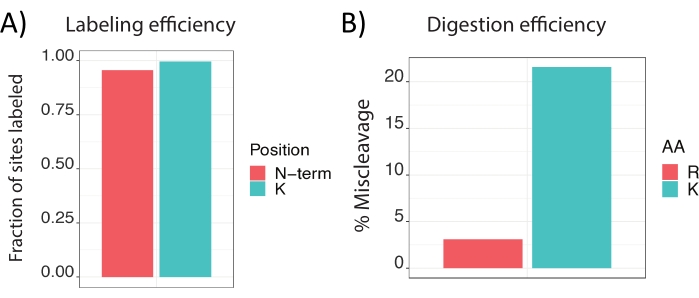
Figure 1: Carrier preparation quality control. DO-MS plots depicting (A) labeling efficiency of a successfully prepared carrier at sites of labeling with TMT: the primary amine on the side chain of lysine and the peptide N-terminus, and (B) the digestion efficiency at amino acid residues of tryptic cleavage. Abbreviation: TMT = tandem mass tag. Please click here to view a larger version of this figure.
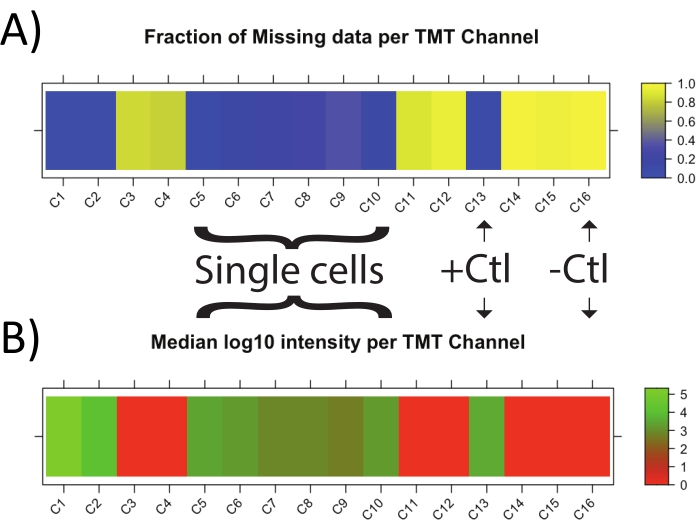
Figure 2: Single-cell preparation quality control. DO-MS plots depicting (A) the fraction of missing data in single cells (C5-10), controls (C13,16), carrier (C1), and reference (C2), and (B) the intensity of single cells and controls. Tags 127C, 128N, 131C, 132N, 133N, and 133C, which correspond to C3, C4, C11, C12, and C15, are unused in this particular set. The positive control consists of cellular material processed to peptides in bulk, then diluted to a level of 2-5 cells/μL prior to labeling. The negative control consists of a well subjected to all preparatory steps used for single cells, just without the addition of a single cell. Abbreviation: TMT = tandem mass tag. Please click here to view a larger version of this figure.
Discussion
One of the keys to successful preparation and analysis of single cells by SCoPE2 is the preparation of the carrier and reference channels. The suggested carrier size is 100 to 200 cells; however, the number of cells needed for a particular single-cell experiment can be determined based on principles as discussed elsewhere21. For the suggested size, at least ~11,275 cells are needed per 384-well plate, allowing for a 200-cell carrier and 5-cell reference in each set. It can be advantageous to isolate more cells if the desired cell populations are not a limiting factor, to simply have extra material in case of mistakes (as was done in this protocol, isolating 22,000 cells instead of 11,275 cells). The amount of carrier and reference needed for the number of desired plates should be prepared in a single batch to minimize any batch-to-batch variation that can cause peptide identification or quantification to differ between batches.
Before isolating single-cells into wells of a 384-well plate, it is important to consider the design of the plate layout. Within each 384-well plate, it is recommended to implement both positive and negative controls. Negative controls are wells in which no cells are added but undergo the same reagent additions and procedures as the single-cell wells. Positive controls are wells in which cell lysate diluted to 2-5 cells/µL is added instead of single cells. This is particularly important to implement when single-cell isolation methods are not well-validated. Each 384-well plate should ideally have a randomized distribution of single cells and controls (e.g., not have negative or positive controls in just one row). Each plate should have an equal distribution of all cell populations of interest, rather than isolating one cell type in one plate and a second cell type in a second plate. This will avoid tying batch effects with cell types of interest. In addition, if more than one 384-well plate is needed for analysis, it is advisable to isolate cells in a single session, rather than spreading the isolation step over several days, if possible.
Lysis of both single cells and carrier/reference takes place in water through a freeze-heat cycle23. It is anticipated that most proteases have been denatured during this step. Trypsin is then added immediately following the denaturation step at a high concentration, many orders of magnitude greater in concentration than even the most abundant cellular proteases. By mass action, most products of protease activity will be due to trypsin.
Reduction/alkylation of cysteine residues is not performed in this protocol before trypsin digestion. Cysteine-containing peptides represent roughly 10% of tryptic peptides from the human proteome. We observe fewer cysteine-containing peptides using an approach without reduction/alkylation. These steps use reagents that are incompatible with labeling by TMT (NHS-ester chemistry) immediately following digestion.
In this TMT labeling strategy, the carrier and references are labeled with 126 and 127N, respectively, while single-cell and control wells are labeled with 128C through 135N. The two labels after the reference, 127C and 128N, are not used due to isotopic cross contamination arising from the carrier and reference channels, which are clearly much more abundant in peptide material than the single cells. In total, there are 12 TMT channels per set that can be used for labeling single cells or control wells with TMTpro-16plex, or 14 TMT channels per set with TMTpro-18plex.
The critical steps in the protocol to perform single-cell proteomics measurements using this protocol include preparation of the carrier, single-cell isolation, lysis, digestion, barcode labeling, and proper mass spectrometry parameters. These steps have been outlined in this document and have been extensively detailed elsewhere17,21,25. Each step has a corresponding plot or set of plots in DO-MS that allow easy quality control. For example, in the case of successful creation of the carrier, plots depicting the number of peptides identified, the labeling efficiency, and the percentage of miscleavage rate allow verification of its successful preparation. Representative DO-MS reports are included in this publication and can be seen at http://scope2.slavovlab.net/ for the original data16. These DO-MS reports allow for assessing optimization of the method in directions not yet investigated by the authors, such as longer LC gradients, different digestion enzymes, or alternative chemical barcodes.
Currently, the serial analysis of peptides by data-dependent acquisition algorithms limits the number of peptides that can be analyzed in an LC run of a reasonable length. This is in part due to the longer fill times required for the successful acquisition of enough ions for reliable single-cell quantification21. An inherent limitation to using the carrier is the lack of direct assessment of digestion and labeling efficiency of the single cells. Currently, one solution to this limitation is performing quality control on a larger sample processed alongside the single cells.
We proposed a number of opportunities for improving single-cell proteomics, such as building mass spectrometers with multiple analyzers. The current method allows the quantification of thousands of proteins from single mammalian cells at a speed of approximately 100 cells per day with commercially available reagents and equipment. SCoPE2, the second generation of the SCoPE-MS approach, significantly improved upon its predecessor with respect to the number of cells analyzable per unit time, the number of proteins analyzable per unit time, the time needed for sample preparation, the accessibility of reagents and equipment, and the overall cost of preparation and analysis per single cell. Membrane-bound proteins are accessible using the freeze-heat lysis and protease digestion, as shown in comparison to urea lysis23. The method identifies many proteins from the top third of the proteome with respect to abundance16. If the modified proteoform is in the top third of the proteome, and the modified peptide is amenable to mass spectrometry analysis (ionizes well, has a mass difference from unmodified peptide), it will more likely be amenable to this protocol. This method may be fruitfully applied in biological systems where there is meaningful heterogeneity amongst cells, such as in differentiation, senescence, or immunological responses (i.e., phagocytosis).
Declarações
The authors have nothing to disclose.
Acknowledgements
We would like to acknowledge the Fall 2021 NSF I-Corps Program (Northeastern University site) award that has funded this publication.
Materials
| 384-well plate, standard PCR plate | Thermo Fisher Scientific | AB1384 | Polypropylene plates should be used, if need to substitute. If substituted, levels of potential polymer contamination from other plates should be assessed prior to SCoPE2 preparation. |
| Acetonitrile (for preparation of buffers), Optima LC-MS/MS grade | Fisher Scientific | A955-1 | This acetonitrile is used for any buffers, namely the 50% acetonitrile that is used to pass through wells after passing through the carrier and reference when combining SCoPE2 sets. Caution: Acetonitrile is a flammable liquid. It can irritate skin, eyes, respiratory tract, and central nervous system. Be sure to wear personal protective equipment. Handle under a chemical fume hood. |
| Acetonitrile (for preparation of TMT ), anhydrous, 99.8% | Sigma Aldrich | 271004-100ML | This acetonitrile is used to resuspend TMT labels at the manufacturer concentration. Caution: Acetonitrile is a flammable liquid. It can irritate skin, eyes, respiratory tract, and central nervous system. Be sure to wear personal protective equipment. Handle under a chemical fume hood. |
| Adhesive PCR plate foils | Thermo Fisher Scientific | AB0626 | |
| Autosampler vial screw thread caps | Thermo Fisher Scientific | C5000-51B | |
| Autosampler vial spinner (i.e. myFuge 5) | MTC Bio | C2595 | This model does not have any speed control. It is simply used to colelct liquid at the bottom of autosampler vials. |
| Benzonase nuclease | Sigma Aldrich | E1014-25KU | |
| Clear glass screw thread vials, 9 mm | Thermo Fisher Scientific | 60180-509 | |
| Formic Acid, Pierce, LC-MS/MS grade | Thermo Fisher Scientific | 85178 | Caution: Formic acid is a flammable liquid. It can cause serious eye damage or skin burns. Be sure to wear personal protective equipment. Handle in a well-ventilated area. |
| Glass autosampler inserts, 9 mm | Thermo Fisher Scientific | C4010-630 | |
| Hydroxylamine (HA), 50% wt/vol | Sigma Aldrich | 467804-50ML | Caution: Hydroxylamine can cause skin irritation. Be sure to wear personal protective equipment. |
| Mantis microfluidic chips, low-volume 3PFE chips | Formulatrix | MCLVPR2 | These chips are meant to be used with the Mantis microfluidics liquid handler to dispense organic solutions. These can be used to dispense TMT labels. |
| Mantis microfluidic chips, low-volume silicone chips | Formulatrix | MCLVS12 | These chips are meant to be used with the Mantis microfluidics liquid handler to dispense aqueous solutions. These cannot be used to dispense TMT labels. |
| Mantis microfluidic liquid handler | Formulatrix | Liquid handler can be substituted. However, it is important to check if the system is compatible with 100% acetonitrile (as in the case of TMT labels), and that the system does not introduce polymer contamination or other type of contamination into the samples. | |
| Mantis PCR plate adapter with wide conical pins for automated plate handling | Formulatrix | 232400 | |
| MassPREP peptide mixture | Waters | 186002337 | Mixture of nine nontryptic peptides |
| PCR tube spinner (i.e., 16-place microcentrifuge for 0.2 mL tubes) | USA Scientific | 2621-0016 | This model does not have any speed control. It is simply used to collect liquid at the bottom of tubes. |
| PCR tubes: TempAssure 0.2 mL PCR 8-tube strips | USA Scientific | 1402-3900 | If substituted, levels of potential polymer contamination from other plates should be assessed prior to SCoPE2 preparation. |
| Phosphate-buffered saline (PBS), 10x, pH 7.4, RNase-free | Thermo Fisher Scientific | AM9625 | |
| Plate spinner (i.e., PlateFuge microcentrifuge) | Benchmark Scientific | Model C2000 | This model does not have any speed control. It is simply used to collect liquid at the bottom of wells. |
| Thermal Cycler (i.e., C1000 Touch with 384-well module) | Biorad | 1851138 | |
| TMTpro 16plex Label Reagent Set, 1 x 5 mg | Thermo Fisher Scientific | A44520 | |
| Triethylammonium bicarbonate (TEAB), 1 M, pH 8.5 | Sigma Aldrich | T7408100ML | |
| Trypsin Gold Mass Spectrometry Grade | Promega | V5280 | This is trypsin is of very high purity. Other trypsin reagents can vary in their levels of purity. Level of purity is important for achieving positive SCoPE2 results. Caution: Trypsin can cause skin, respiratory, and eye irritation. Be sure to wear personal protective equipment. Handle under a chemical fume hood. |
| Vortex (Analog vortex mixer) | VWR | Model 58816-121 | |
| Water bath sonicator (i.e., 2.8 L ultrasonic cleaner with digital timer) | VWR | 97043964 | |
| Water, Optima LC-MS/MS grade | Fisher Scientific | W6-1 | All solutions that need to be diluted or made are recommended to be made with mass-spectrometry grade water. Any dilutions and/or master mixes should be made fresh. Contamination resulting from lower-grade water can negatively affect peptide detection and single-cell data quality . |
Referências
- Symmons, O., Raj, A. What’s luck got to do with it: single cells, multiple fates, and biological nondeterminism. Molecular Cell. 62 (5), 788-802 (2016).
- Paul, I., White, C., Turcinovic, I., Emili, A. Imaging the future: the emerging era of single-cell spatial proteomics. The FEBS Journal. 288 (24), 6990-7001 (2020).
- Levy, E., Slavov, N. Single cell protein analysis for systems biology. Essays in Biochemistry. 62 (4), 595-605 (2018).
- Nagasawa, S., Kashima, Y., Suzuki, A., Suzuki, Y. Single-cell and spatial analyses of cancer cells: toward elucidating the molecular mechanisms of clonal evolution and drug resistance acquisition. Inflammation and Regeneration. 41 (1), 22 (2021).
- Shaffer, S. M., et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 555 (7695), 274 (2018).
- Pan, D., Jia, D. Application of single-cell multi-omics in dissecting cancer cell plasticity and tumor heterogeneity. Frontiers in Molecular Biosciences. 8, 757024 (2021).
- Segerstolpe, &. #. 1. 9. 7. ;., et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metabolism. 24 (4), 593-607 (2016).
- Tritschler, S., Theis, F. J., Lickert, H., Böttcher, A. Systematic single-cell analysis provides new insights into heterogeneity and plasticity of the pancreas. Molecular Metabolism. 6 (9), 974-990 (2017).
- Hanna, S. J., Tatovic, D., Thayer, T. C., Dayan, C. M. Insights from single cell rna sequencing into the immunology of type 1 diabetes- cell phenotypes and antigen specificity. Frontiers in Immunology. 12, 751701 (2021).
- Baron, M., et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Systems. 3 (4), 346-360 (2016).
- Franks, A., Airoldi, E., Slavov, N. Post-transcriptional regulation across human tissues. PLoS Computational Biology. 13 (5), 1005535 (2017).
- Slavov, N. Unpicking the proteome in single cells. Science. 367 (6477), 512-513 (2020).
- Specht, H., Slavov, N. Transformative opportunities for single-cell proteomics. Journal of Proteome Research. 17 (8), 2565-2571 (2018).
- Slavov, N. Learning from natural variation across the proteomes of single cells. PLoS Biology. , (2021).
- Slavov, N. Driving single cell proteomics forward with innovation. Journal of Proteome Research. 20 (11), 4915-4918 (2021).
- Specht, H., et al. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biology. 22 (1), 50 (2021).
- Petelski, A. A., et al. Multiplexed single-cell proteomics using SCoPE2. Nature Protocols. 16 (12), 5398-5425 (2021).
- Slavov, N. Scaling up single-cell proteomics. Molecular and Cellular Proteomics: MCP. 21 (1), 100179 (2022).
- Cong, Y., et al. Ultrasensitive single-cell proteomics workflow identifies> 1000 protein groups per mammalian cell. Chemical Science. 12 (3), 1001-1006 (2021).
- Lombard-Banek, C., et al. In vivo subcellular mass spectrometry enables proteo-metabolomic single-cell systems biology in a chordate embryo developing to a normally behaving tadpole (X. laevis). Angewandte Chemie. 60 (23), 12852-12858 (2021).
- Specht, H., Slavov, N. Optimizing accuracy and depth of protein quantification in experiments using isobaric carriers. Journal of Proteome Research. 20 (1), 880-887 (2020).
- Budnik, B., Levy, E., Harmange, G., Slavov, N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology. 19 (1), 161 (2018).
- Specht, H., Harmange, G., Perlman, D. H., Emmott, E. Automated sample preparation for high-throughput single-cell proteomics. BioRxiv. , (2018).
- Singh, A. Towards resolving proteomes in single cells. Nature Methods. 18 (8), 856 (2021).
- Huffman, R. G., Chen, A., Specht, H., Slavov, N. DO-MS: Data-driven optimization of mass spectrometry methods. Journal of Proteome Research. 18 (6), 2493-2500 (2019).
- Chen, A. T., Franks, A., Slavov, N. DART-ID increases single-cell proteome coverage. PLoS Computational Biology. 15 (7), 1007082 (2019).
- Cheung, T. K., et al. Defining the carrier proteome limit for single-cell proteomics. Nature Methods. 18 (1), 76-83 (2021).
- Leduc, A., Huffman, R. G., Slavov, N. Droplet sample preparation for single-cell proteomics applied to the cell cycle. bioRxiv. , (2021).
- Vanderaa, C., Gatto, L. Utilizing Scp for the analysis and replication of single-cell proteomics data. bioRxiv. , (2021).
