Culturing and Genetically Manipulating Entomopathogenic Nematodes
Summary
Entomopathogenic nematodes live in symbiosis with bacteria and together they successfully infect insects by undermining their innate immune system. To promote research on the genetic basis of nematode infection, methods for maintaining and genetically manipulating entomopathogenic nematodes are described.
Abstract
Entomopathogenic nematodes in the genera Heterorhabditis and Steinernema are obligate parasites of insects that live in the soil. The main characteristic of their life cycle is the mutualistic association with the bacteria Photorhabdus and Xenorhabdus, respectively. The nematode parasites are able to locate and enter suitable insect hosts, subvert the insect immune response, and multiply efficiently to produce the next generation that will actively hunt new insect prey to infect. Due to the properties of their life cycle, entomopathogenic nematodes are popular biological control agents, which are used in combination with insecticides to control destructive agricultural insect pests. Simultaneously, these parasitic nematodes represent a research tool to analyze nematode pathogenicity and host anti-nematode responses. This research is aided by the recent development of genetic techniques and transcriptomic approaches for understanding the role of nematode secreted molecules during infection. Here, a detailed protocol on maintaining entomopathogenic nematodes and using a gene knockdown procedure is provided. These methodologies further promote the functional characterization of entomopathogenic nematode infection factors.
Introduction
Research on entomopathogenic nematodes (EPN) has intensified over the past few years due primarily to the utility of these parasites in integrated pest management strategies and their involvement in basic biomedical research1,2. Recent studies have established EPN as model organisms in which to examine the nematode genetic components that are activated during the different stages of the infection process. This information provides critical clues on the nature and number of molecules secreted by the parasites to alter host physiology and destabilize the insect innate immune response3,4. Simultaneously, this knowledge is commonly supplemented by novel details on the type of insect host immune signaling pathways and the functions they regulate to restrict the entry and spread of the pathogens5,6. Understanding these processes is crucial for envisioning both sides of the dynamic interplay between EPN and their insect hosts. Better appreciation of EPN-insect host relationship will undoubtedly facilitate similar studies with mammalian parasitic nematodes, which can lead to the identification and characterization of infection factors that interfere with the human immune system.
The EPN nematodes Heterorhabditis sp. and Steinernema sp. can infect a wide range of insects, and their biology has been intensely studied previously. The two nematode parasites differ in their mode of reproduction with Heterorhabditis being self-fertilized and Steinernema undergoing amphimictic reproduction, although recently S. hermaphroditum was shown to reproduce by self-fertilization of hermaphrodites or through parthenogenesis7,8,9. Another difference between Heterorhabditis and Steinernema nematodes is their symbiotic mutualism with two distinct genera of Gram-negative bacteria, Photorhabdus and Xenorhabdus, respectively, which are both potent pathogens of insects. These bacteria are found in the free-living and non-feeding infective juvenile (IJ) stage of the EPN, which detect susceptible hosts, gain access to the insect hemocoel where they release their associated bacteria that replicate rapidly, and colonize insect tissues. Both the EPN and their bacteria produce virulence factors that disarm insect defenses and impair homeostasis. Following insect death, the nematode IJs develop to become adult EPN and complete their life cycle. A new cohort of IJs formed in response to food deprivation and overcrowding within the insect cadaver finally emerges in the soil to hunt suitable hosts9,10,11,12.
Here, an efficient protocol for maintaining, amplifying and genetically manipulating EPN nematodes is described. In particular, the protocol outlines the replication of symbiotic H. bacteriophora and S. carpocapsae IJs, the generation of axenic nematode IJs, the production of H. bacteriophora hermaphrodites for microinjection, the preparation of the dsRNA, and the microinjection technique. These methods are essential for understanding the molecular basis of nematode pathogenicity and host anti-nematode immunity.
Protocol
1. Production of symbiotic nematode infective juveniles
- Cover a Petri dish (10 cm) with a piece of filter paper and add approximately 10-15 Galleria mellonella larvae (Figure 1A).
- Using a pipette, dispense 2 mL of water containing about 25-50 IJs per 10 µL suspension onto the waxworms. Store the Petri dish in a cabinet at room temperature.
- Depending on the moisture of the filter paper, add 1-2 mL of water every 2 days. Waxworms infected with IJs normally will die within 48 h.
- Prepare White's water traps approximately 10 days after the waxworms are infected with the IJs8 (Figure 1B,C). Carefully transfer the dead insects onto the unsoaked part of the filter paper. Use tap water to fill the bottom Petri dish and place a cap of a 15 mL tube as a spacer for ventilation.
- Transfer the soft and fragile waxworms carefully by lifting them slowly from the filter paper using a pair of plastic forceps. Use tap water instead of demineralized or deionized water. The latter cause nematode aggregation.
- Make sure the water level in the water trap reaches approximately half the height of the Petri dish. Expect that water level will change over time depending on temperature and humidity conditions in the room.
- When the water in the small Petri dish becomes cloudy due to the presence of nematodes, use a pipette to move the new generation of IJs into a T25 or T75 cell culture flask.
- Add tap water up to about 40% of the volume until the appropriate density is reached (Figure 1C). Avoid nematode congestion and store the cell culture flasks horizontally.
- Add more water to the bottom Petri dish and repeat steps 1.6 and 1.7 until IJs stop emerging from the insect carcasses in approximately 3-5 days.
2. Production of axenic nematode infective juveniles
NOTE: Axenic nematodes are used because after the nematode-bacteria complex dissociates inside the insect, each mutualistic partner elicits a distinct host immune response5. The mutant strain Ret16 of Photorhabdus temperata is used because these bacteria support the growth of H. bacteriophora but fail to colonize the nematode gut13,14.
- Heterorhabditis bacteriophora infective juveniles
- Using a sterile pipette tip or spatula, scrape several flakes of a frozen culture of P. temperata Ret16 onto a MacConkey plate and streak for single colonies. Incubate the plate for 2-3 days at 28 °C.
- Inoculate 10 mL of LB broth with a colony in a 50 mL tube and incubate the culture overnight at 28 °C in a shaking incubator. Use only the primary phase colonies that are red on MacConkey agar. The secondary-phase colonies will be off-white on MacConkey agar.
- Wash 100 µL of overnight culture with 900 µL of 1x PBS by centrifuging in a 1.5 mL microcentrifuge tube at 17,900 x g. Decant the supernatant. Dilute the culture 10x in 1x PBS. Leave the tube on ice.
- Immerse the waxworms into a 70% ethanol solution, dry the insects with a paper towel, and place them in a 50 mL tube.
- Place the tube on ice for 20 min to immobilize the waxworms.
- Use a filter paper to cover the top and bottom halves of a Petri dish (10 cm). Use the Petri dish lid covered with filter paper as a base support for injection. Moisten the filter paper of the bottom Petri dish and transfer on ice to help the injected waxworms recover.
- Pipette 50 µL of ice-cold bacteria on a piece of parafilm and prepare the 22 G needle syringe. Press the plunger gently to remove the air at the tip of the needle.
- Keep a waxworm close to the posterior end under the stereoscope (Figure 2).
- Inject 50 µL of the bacterial culture into the dorsal side of the thorax, preferably at the junction between two segments. To minimize internal damage, inject just underneath the cuticle, as parallel to the waxworm as possible. It is natural for a droplet of hemolymph to bleed out when the waxworm is pierced
- Transfer the injected insects to the recovery Petri dish.
- Repeat steps 2.1.7-2.1.9 until all insects are injected with the bacteria. To anesthetize the insects, place them on ice for 5 min.
- Place the Petri dish in the dark (e.g., drawer or cabinet) and add tap water to the filter paper if it appears dry. Waxworms will succumb 2 days after injection, and appear brick red after approximately 3-4 days. If the insects appear brown, the bacterial infection has been unsuccessful.
- At 7 days post infection, transfer the insects with the characteristic brick red color onto a fresh filter paper-lined Petri dish, and continue with "Production of symbiotic nematode infective juveniles" from step 1. If using symbiotic nematodes, repeat the procedure from step 2.1. using the newly produced IJs from the first round.
- Surface-sterilizing H. bacteriophora IJs
- Collect sufficient symbiotic H. bacteriophora and candidate axenic IJs by centrifugation to make a 100 µL pellet in a 1.5 mL centrifuge tube.
- Add 500 µL of 5% bleach to the pellet in each microcentrifuge tube and invert. Incubate for 10 min. Centrifuge at 17,900 x g for 1 min. Remove the supernatant.
- Wash the pellet with 1 mL of sterile water and repeat centrifugation. Remove the supernatant. Repeat this step four more times.
- Verifying axenicity
- Pipette 400 µL of sterile water to the washed symbiotic and candidate-axenic H. bacteriophora nematodes.
- Place the nematodes over the insects in the Petri dish and label the treatments accordingly.
- Place the Petri dishes with the infected insects in the dark and check periodically whether the waxworms turn red.
NOTE: Waxworms infected with H. bacteriophora symbiotic nematodes will become red within 2 days of infection. Waxworm discoloration is due to the secretion of various compounds produced by mutualistic Photorhabdus bacteria during the infection process. Lack of red color in the infected waxworms 4 days after infection confirms that the H. bacteriophora are axenic. This is because P. temperata Ret16 bacteria are not present in the nematode gut.
- Alternative method for verifying axenicity
- Surface-sterilize symbiotic H. bacteriophora IJs and candidate-axenic H. bacteriophora IJs as described in step 2.1.14.
- Homogenize approximately 500 IJs in 500 µL of sterile 1x PBS in microfuge tubes using sterile pestles. Spin down the homogenate, decant the supernatant, and spread it onto LB agar. Incubate the plates at 28 °C for 24 h.
- Count the colony forming units (CFU) for each plate. Samples from axenic H. bacteriophora will not form any colonies of the symbiotic P. luminescens bacteria.
- Steinernema carpocapsae Infective Juveniles
- Preparation of lipid agar plates (300 mL solution makes approximately 20 split plates)
- Weigh 2.4 g of Nutrient Broth, 4.5 g of yeast extract, and 1.5 g of agar and add them to 267 mL of deionized water. Autoclave the solution.
- Add 3 mL of 1 M MgCl2, 1.2 mL of corn oil and 28.8 mL of 7% corn syrup.
- Prepare and add to the solution 300 µL of 30 mg/mL kanamycin and 300 µL of 50 mg/mL ampicillin (sterilized with 0.2 µm filter).
- Decant the mix onto one half of a divided Petri dish/split plate.
- Preparation of X. nematophila (strain ΔrpoS) bacterial lawn
- Culture X. nematophila (Xn) ΔrpoS directly from a frozen bacterial stockin 2 mL of LB/kan/amp solution on a shaker at 30 °C overnight.
- Inoculate 5 mL of LB medium containing 30 µg/mL kanamycin and 50 µg/mL ampicillin with 250 µL of the overnight culture and grow the bacteria overnight on a shaker.
NOTE: Xn bacteria do not grow well when streaked directly on selection lipid agar media from a frozen stock. If necessary, primary and secondary phase Xn can be firstly confirmed on NBTA media (nutrient agar supplemented with 25 mg of bromothymol blue l−1 and 40 mg triphenyltetrazolium chloride l−1) before starting a fresh liquid culture. - Pipette 100 µL of the culture on the lipid agar plates and spread on the entire plate with a sterile inoculating loop. Incubate the plates for 24 h at 30 °C.
- Surface-sterilizing S. carpocapsae IJs
- Leave a flask containing IJs at an angle such that the IJs sink to a corner of the flask. Aspirate 1 mL of the settled IJs into a microcentrifuge tube. Centrifuge at 17,900 x g for 10 s. Remove the supernatant.
- Add 1 mL of 1% freshly prepared bleach solution. Invert tubes to mix thoroughly. Incubate for 1 min (prolonged bleach incubation will lead to IJ death). Spin again for 10 s. Remove supernatant.
- To remove bleach residue, wash the nematodes with 1 mL of sterile distilled water and centrifuge for 10 s. Remove supernatant. Repeat the wash four more times.
- Estimate the IJ count per mL by counting 10-20 µL of the washed IJs under the stereoscope and adjust the concentration of the IJs accordingly.
- Rearing S. carpocapsae IJs
- Transfer with a pipette around 1000 IJs onto the lipid agar split plates (Figure 3, left image). Place the plates in a humidified cabinet or drawer. Use damp paper towels to increase moisture. Maintain the plates at room temperature (22-25 °C).
- Check the paper towels every other day to make sure they stay moist. If necessary, add water every other day to the paper towels to maintain humidity in the cabinet or drawer. Alternatively, place a water bath to the bottom of the cabinet to increase humidity.
- When IJs only appear, add water to the other side of the plate, and place a layer of filter paper across the middle of the plate (water trap, Figure 3, right picture). Collect this water containing the first round IJs into a T75 cell culture flask. These are the Round I S. carpocapsae nematodes.
- Surface-sterilize with bleach (step 2.2.3) again and repeat the process from step 2.2 using the Round 1 S. carpocapsae nematodes. The result will be Round 2 S. carpocapsae nematodes.
- Check under a stereoscope every few days to track the development of the nematodes.
- Verifying the axenic state of S. carpocapsae IJs
- Surface-sterilize the Round 1, Round 2, and symbiotic S. carpocapsae IJs with bleach as described in step 2.2.3.
- Homogenize approximately 400-700 IJs in microfuge tubes using sterile plastic pestles. Centrifuge the homogenate, decant the supernatant, and spread the solution onto LB agar plates. Keep the agar plates in an incubator set at 28 °C for 24 h.
- Count the formation of CFU for each plate. Samples from axenic nematodes will not produce any bacterial growth.
- Verifying the axenic state of S. carpocapsae IJs by PCR
- Surface-sterilize the Round 1, Round 2, and symbiotic S. carpocapsae IJs with bleach as described in step 2.2.3.
- Extract DNA from the homogenate. A detailed extraction procedure is described in Section 4.1.2 below.
- Conduct PCR using the XptA2 primers with annealing temperature 61 °C:
XptA F: 5′-GCCTGGAAAGAGTGGACGAA-3′.
XptA R: 5′-GTAAGACCAAGGGGCACTCC-3′.
NOTE: These primers amplify the insecticidal gene XptA2 of X. nematophila, producing a 231 bp size amplicon. The cycling program was as follows: 95 °C for 2 min, 34 cycles of 95 °C for 30 s, annealing temperature of 61 °C for 1 min and 73 °C for 1 min followed by 72 °C for 10 min. - Visualize the amplified fragments by separation in an 1.5% agarose gel. Axenic surface-sterilized samples will not form any bands.
- Preparation of lipid agar plates (300 mL solution makes approximately 20 split plates)
3. Raising H. bacteriophora hermaphrodites for microinjection
- Prepare 6 cm Petri dish plates with NA+chol (1.5x Nutrient Broth, 1.5% agar, and 10 µg/mL cholesterol).
- Prepare 3 mL of PP3 solution (2% Protease Peptone #3, PP3) in a 10 mL culture tube.
- Use a sterile 20 µL tip to scrape the top of the P. luminescens glycerol stock (25% vol/vol sterile glycerol) maintained at -80 °C and drop it in the culture tube.
- Incubate the tube overnight in a temperature-controlled shaker set at 28 °C, 200 rpm.
- On the following day, the culture will be light red in color. Plate 50 µL of the culture on a 6 cm NA+chol Petri dish and spread it using a bacterial spreader in a circular manner.
- Place the plates in a 28 °C incubator. The P. luminescens lawn will be ready in 24-36 h and will appear light red in color.
- Inoculate the plate with 50-100 surface sterilized IJs and maintain the plate at 28 °C.
- Healthy L4 hermaphrodite will start appearing approximately 48-54 h after inoculation. Healthy, non-starved late L4 H. bacteriophora hermaphrodites are required for injection.
4. Preparation of dsRNA
- Primer design
- Design primers for dsRNA to target ~500 base pair exon regions of H. bacteriophora DNA.
- Primers for regions of interest can be determined using Primer3 (https://primer3.ut.ee) or a similar program, selecting for an optimum product length of 500 base pairs, Tm of 60 °C, and primer length of 22 nucleotides.
- Add a T7 site (TAATACGACTCACTATAGGG) to the 5′ ends of each forward primer to allow for in vitro transcription.
- Design primers for dsRNA to target ~500 base pair exon regions of H. bacteriophora DNA.
- Genomic DNA Isolation
- Use genomic DNA isolated from frozen pellets of ~50,000 H. bacteriophora IJs for dsRNA synthesis.
- Use an IJ pellet resuspended in 50 µL of lysis buffer (50 mM KCl, 0.05% (w/v) gelatin, 10 mM Tris-HCl pH 8.2, 0.45% Tween 20, 60 µg/mL Proteinase K, 2.5 mM MgCl2) placed at −80 °C for at least 30 min.
- Homogenize the pellet with a small tube pestle.
- Warm the solution to room temperature and incubate at 60 °C for 2 h, vortexing every 15 min.
- Denature the Proteinase K by incubating the homogenized tissue for 15 min at 95 °C.
- Cool the sample to 4 °C and centrifuge at 3,400 x g for 1 min.
- Use the resulting supernatant as a template for subsequent PCR.
- Set up a 50 µL PCR reaction using a commercial mastermix with 200 ng of template DNA, 0.2 µM of forward and reverse primer, and the manufacturer's suggested cycling conditions.
- Analyze the PCR reaction on a 1.2% agarose gel to verify that the reactions produced single bands of the predicted size.
- dsRNA synthesis
- Use a commercial transcription kit.
- Use a 5 µL PCR reaction for in vitro transcription. Follow the manufacturer's instructions.
- Incubate the reaction for 16 h at 37 °C.
- Clean the in vitro transcription reactions with a commercial kit using ammonium acetate/ethanol precipitation to concentrate the dsRNA.
- Suspend the pelleted dsRNA in 10 µL of RNase-free water.
- Quantify the RNA using a spectrophotometer.
- Assess the quality by separating the dsRNA on a 1.2% agarose gel
5. Microinjection
NOTE: An injection pad is a glass coverslip with a layer of 2% (w/v) agarose on the center. When the worms to be injected are transferred to these pads, the agarose layer will immobilize them for the procedure. Normally, extra pads are kept near the microscope for general use.
- Preparing the injection pad
- Boil 2% agarose in water by adding 0.2 g of agarose to 10 mL of water.
- Place 1-4 drops (~50 µL) onto the center of a 50 mm x 70 mm thin glass coverslip.
- Use another coverslip to flatten the drop.
- Let the coverslip dry for 2-3 min before sliding the coverslip off sideways.
- Mark with an 'R' the right side of the upward-facing coverslip.
- Let the slides dry at room temperature for 1 day before using them.
- Preparation of injection needles
- Use 1.0 mm glass capillaries containing an internal fine filament. This allows efficient filling of the needle.
NOTE: Any commercial needle puller (e.g., Narishige PB-7) can be used. - Turn the needle puller on.
- Make sure the heater is set to max and the solenoid is set to 8.40 to ensure required needle sharpness and proper length.
- Insert the capillary tube into the top knob ridge through the filament and tighten the top knob. The top of the capillary tube will be level with the top of the needle puller and the heating filament will be in the middle of the capillary.
- Move the bottom unit to the top and then tighten the bottom knob. Make sure that the capillary is securely placed.
- Close the cover of the needle puller and press start. The green light will turn on. After a few minutes the capillary will be heated and pulled apart to separate in two halves.
NOTE: The two separated needles are not of the same length, but they can both be used for the injections. Longer needle points are preferred. - Arrange the needles in a vertical position with their points downward using a piece of modeling clay. Alternatively, store the needles in a Petri dish held in place by two strips of modeling clay.
- Use 1.0 mm glass capillaries containing an internal fine filament. This allows efficient filling of the needle.
- Loading the needle
NOTE: The nucleic acid solution must be centrifuged for at least 10 min at 20,784 x g to pellet impurities that may be present in the suspension. The centrifugation of the impurities keeps them from blocking the flow of the injection needle.- Use either of the two methods to load nucleic acids in the needles.
- Method 1
- Use a pipette to transfer approximately 0.5 µL of the centrifuged DNA sample to the unpulled end of the injection needle. This will appear as a tiny drop of liquid sitting on top of the capillary tube.
- Stand by for 5 to 7 min to allow the liquid sample to occupy the end of the needle. The final product will be a filled needle with no air bubbles present.
- Use a dissection microscope to confirm the presence of solution in the needle tip before injection.
- Method 2
- Use loading tips for the microinjector.
- Using this loading tip, pipette 5 µL of the nucleic acid solution.
- While the pulled needle is on the needle puller, loosen the top knob, move the upper pulled needle slightly upwards, and retighten the knob.
- Carefully insert the tip of the loader into the capillary tube and extend it to the bottom of the injection needle.
- Pipette the nucleic acid solution into the injection needle.
- Remove the loader from the pulled needle and keep it separate for further loading, if required. Be sure to switch out loaders when loading a different nucleic acid mix.
- Preparing the injection microscope
NOTE: An inverted microscope with diffused illumination from the microscope base is used for worm preparation, microinjection, and recovery. The use of a high-resolution inverted microscope with 10x and 40x objectives is described. The microscope is fitted with a needle manipulator which holds the needle and brings it into position for the injection. Microinjection oil is used to prevent the worm from rapid dehydration while on the agarose layer of the injection pad. The suggested oil to use is Halocarbon Oil 700.- Switch on the lamp and calibrate the brightness with the control panel.
- Check the number on the condenser Wollaston prism; it will correspond to the used objective (40x).
- Confirm that the DIC turret, marked by "DIC", is at the right position.
- Ensure that the knob on the objective is set at zero (0).
- Turn the Ph ring to the right until the end.
- Adjust the polarizer in a way that the arrow is at horizontal position.
- Put a piece of paper on the stage to observe the focal point (light).
- Shut the field diaphragm to the point that there is a spot on the paper.
- Use the dial on the left to move the condenser up and down to the point that a very sharp light spot on the piece paper is seen.
- Open up the field diaphragm slowly up to the point that a small circle of light is visible.
- Take the piece of paper away.
- Make sure that the light spot is at the center of the objective. Otherwise, line up the light spot with the center of the objective by adjusting the silver screws on the top of the condenser.
- Fully open the field diaphragm.
- Check that the analyzer ICT/P is pressed in.
- Mounting the injection needle on the microscope
- Turn on the nitrogen gas flow to the microinjection needle. The gas pressure to the needle will be approximately 20 psi.
- Check the micromanipulator knobs (the part of the microscope that moves the needle) are positioned to the center, which allows for the widest range of motion when the needle is mounted.
- Unscrew the assembly that holds the metal rod to remove it from the needle holder.
- Insert the new needle into the top part of the microscope; then install the rubber gasket to keep the needle in position.
- Secure the bottom part of the needle holder by fitting it to the assembly.
- Put the needle into the groove on the micromanipulator and screw it properly to keep it steady. Make sure the screw is approximately 1 in from the end of the micromanipulator.
- Place the tip of the needle in the middle of the visual field at about a 45° angle in relation to the stage plate, using the knobs and coarse controls from the micromanipulator. It is critical to leave the needle tip high enough so that it does not break by accident.
- Breaking the needles
- Place the needle in the micromanipulator where the tip of the needle will normally break to allow the nucleic acid solution to flow through it.
- Break a capillary and place it on top of a glass slide carrying a drop of microinjection oil.
- Place the slide on the microscope and focus.
- Bring the tip of the needle to the same level as the capillary's side using the fine controls of the microscope (Figure 4).
- Turn on the nitrogen flow briefly using the foot actuator to confirm that the tip of the needle is not broken. A quick on-off will suffice.
- Move the needle gently so that it barely touches the capillary's side.
- Tap the side of the microscope lightly to break the needle tip.
- Remove the needle from the capillary and turn on the nitrogen flow to check that the needle is broken properly.
- The needle will create a small injection drop approximately five times larger than the tip of the needle (Figure 4). If the injection drop is of smaller size, break the tip further. If the injection droplet is of larger size, try to use a fresh needle.
6. Microinjection
- Pipette a drop of microinjection oil on an injection pad.
- Dip a flame-sterilized pick into the microinjection oil drop and collect a young H. bacteriophora adult nematode from the plate.
- Pick the nematodes from parts of the plate which are not close to the bacterial lawn to prevent the transfer of high numbers of bacterial cells to the injection pad. Also, harvest a nematode with well-formed gonads.
- Move the nematodes to the injection pad and position them vertically so that the gonads face the needle. Make sure that the vulva points to the same direction as the injection needle, and the two distal gonad arms are in the opposite direction and up against the nematode body wall.
NOTE: Like Pristionchus pacificus, the gonad of H. bacteriophora migrates dorsally from the ventral vulval position and then migrates back around to the ventral position. - Check that the amount of oil is sufficient to prevent nematode desiccation without allowing the nematode to wander around.
- Bring the nematode into focus with the 10x objective by holding the needle well above the slide. In oil, the nematode gonads are viewed as two clear regions close to the anterior and posterior of the worm. Ensure that the gonads are in focus and not the other parts of the worm.
- Arrange the nematode at an angle of 15° to 45° towards the injection needle, which will give more space to the needle inside the gonad. Also, this approach prevents the needle from passing through the entire nematode body when the needle is introduced inside the gonad.
- Position the injection needle and the nematode to the same level by gradually lowering the needle until it is brought to focus (Figure 5). Make sure that as the needle is lowered, it remains next to the nematode and not above the worm.
- Use the 40x objective to focus on the syncytial gonad arm of the nematode.
- Move the needle up and down using the fine adjuster until the tip is in focus together with the nematode syncytial gonad arm.
- Move the nematode slowly toward the needle using the sliding stage. Then gently push the gonad to position with the needle against the nematode body wall, which will result in efficient needle penetration inside the nematode body.
- Briefly step on the foot actuator to start the flow of the nucleic acid solution. A quick on-off is sufficient. If the needle is inside the gonad, the gonad will fill with the solution and swell.
- Once the needle is ensured in the correct position, fill the gonad with the nucleic acid solution until the arms of the gonad are filled. It is important to avoid the expulsion of liquid from the worm through the hole where the needle punctured it.
- Using the sliding stage, carefully move the worm away from the needle.
- Lift the needle and turn it counterclockwise.
- Place a drop of 1x PBS buffer on top of the worm to make it float.
- Use the flame-sterilized pick to lift the worm and place it into another drop of M9 on a fresh P. luminescens seeded plate to rid the worm of excess oil.
- Remove the nematode from the M9 and place it to the far side of the bacterial lawn.
- Use the same plate to transfer several injected nematodes.
- In 2-3 days, evaluate the first generation (F1) nematodes for the transgenic phenotype. Separate the transgenic F1 nematodes onto individual plates to determine which worms will generate stable transgenic lines.
Representative Results
To assess the status of H. bacteriophora nematodes that have gone through the axenization, the presence or absence of P. luminescens bacterial colonies in IJs was determined. To do this, a pellet of approximately 500 IJs that had been previously surface sterilized and homogenized in PBS was collected. The positive control treatment consisted of a pellet of approximately 500 IJs from the nematode culture containing symbiotic P. luminescens bacteria. The pellets of axenized and positive control nematodes were homogenized in 1x PBS. The homogenates were pipetted onto agar and spread to facilitate the growth of individual colonies. The agar plates were incubated at 28 °C for 24 h. The next day, the occurrence of P. luminescens colonies (CFU) was observed. As expected, H. bacteriophora from the stock culture carried symbiotic P. luminsecens cells; however, nematodes devoid of their associated P. luminecens bacteria were axenic and stored separately for future experimentation (Figure 6).
Knockdown of nol-5 gene expression was used as an example to show the efficacy of RNAi using the microinjection process. Microinjection was performed in the parental generation and the effect was observed in the F1 progeny. Knockdown of nol-5 using RNAi microinjection resulted in absence of germline in the gonad of 60% of the progeny (Figure 7).
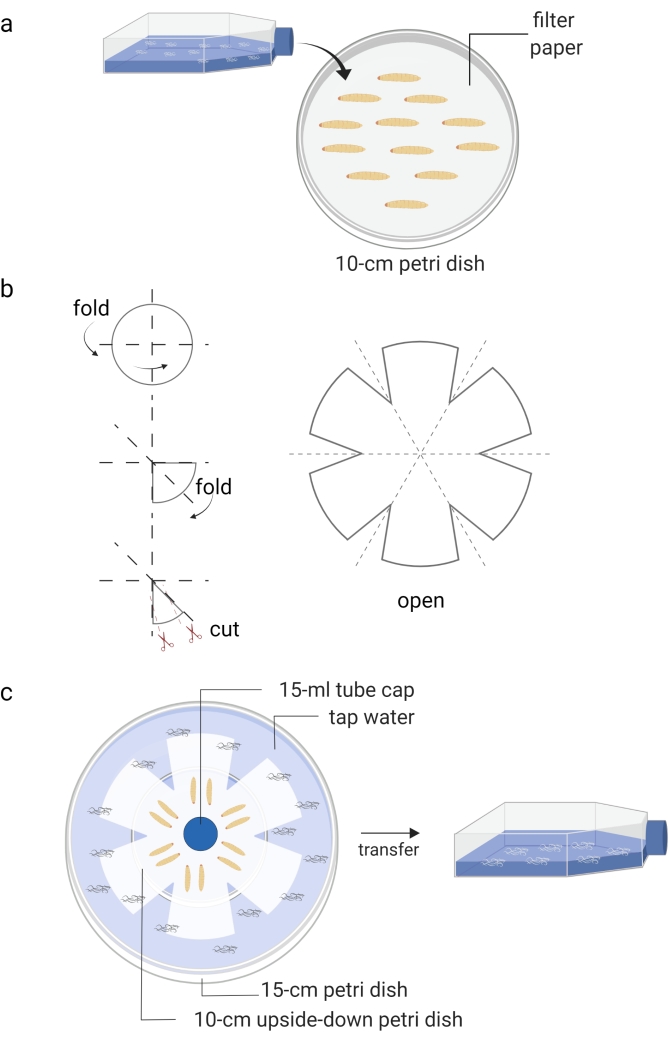
Figure 1: Generation of infective juveniles (free living stage of entomopathogenic nematodes). (a) Infection of Galleria mellonella insect larvae (kept in Petri dish) with nematode infective juveniles (kept in tissue culture flask). (b) Folding of a piece of filter paper for preparation of a water trap. (c) The arrangement of a water trap of entomopathogenic nematodes with dead Galleria mellonella insect larvae containing infective juveniles (12 days post nematode infection). The new generation of infective juveniles will leave the dead insects and move to the water, which is then stored into a tissue culture flask. Images are made using BioRender graphic software (https://biorender.com). Please click here to view a larger version of this figure.
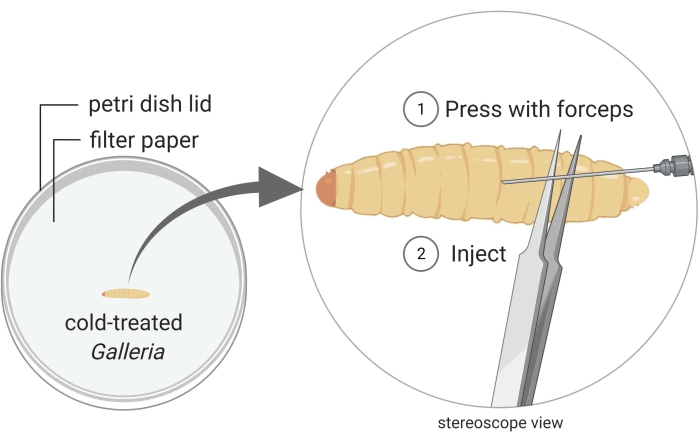
Figure 2: Injection of Galleria mellonella insect larvae with the bacterium strain Photorhabdus temperata Ret16. Waxworms are kept in a Petri dish, and they are immobilized through cold treatment (i.e., they are placed on ice for a few minutes). The posterior part of the insect body is pressed with a pair of forceps to create a swollen surface which is suitable for injection. The injection angle is shallow to prevent injuring internal insect tissues, which would lead to insect death due to careless handling. Images are made using BioRender graphic software (https://biorender.com). Please click here to view a larger version of this figure.
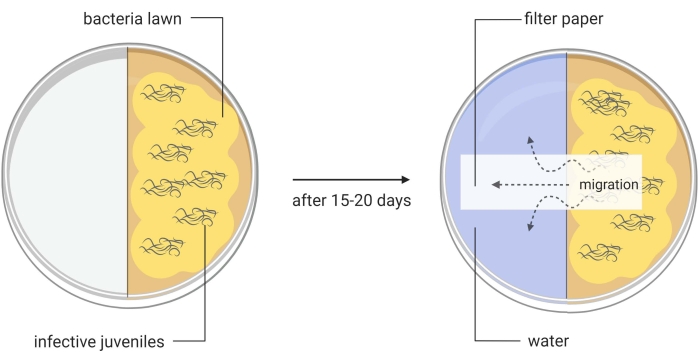
Figure 3: Generation of axenic Steinernema carpocapsae entomopathogenic nematodes. One half of a divided Petri dish contains a lawn of Xenorhabdus nematophila ΔrpoS bacteria on lipid agar and infective juveniles of S. carpocapsae nematodes. This in vitro method induces the infective juveniles to become adult nematodes, which will later produce eggs that will hatch to give rise to the new generation of the parasites. When a large number of newly generated S. carpocapsae infective juveniles appear in this part of the divided Petri dish, water is added to the other half of the dish together with a piece of filter paper to facilitate the migration of the nematodes. Images are made using BioRender graphic software (https://biorender.com) Please click here to view a larger version of this figure.
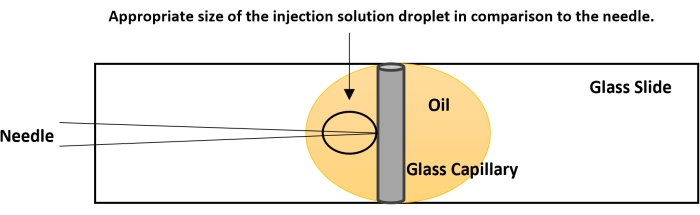
Figure 4: Breaking the needle tip. The tip of the needle is usually closed. The proper flow of the nucleic acid solution is checked before performing the actual injection. On a glass slide, place a drop of halocarbon oil. Place a capillary tube, used for making the needle, vertically as shown in the figure. Bring the capillary to focus at 40x and gently bring the needle down in plane with the capillary. Gently touch the needle tip to the capillary. This creates an opening for the smooth flow of the dsRNA. If no fluid is coming out, perform a quick on-off nitrogen flow using the foot actuator. The pressure from the nitrogen and the contact of the needle tip with the capillary will break the needle. Please click here to view a larger version of this figure.
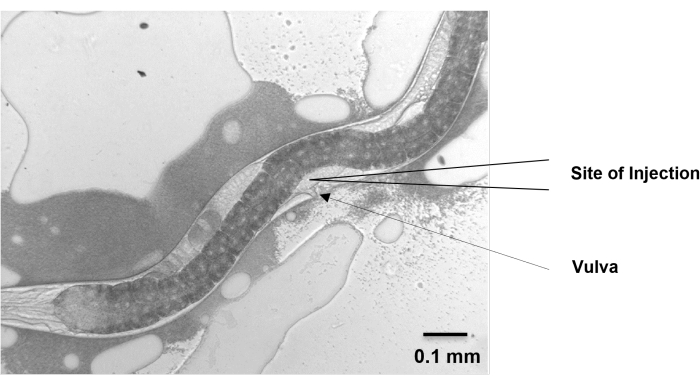
Figure 5: Site of injection. The gonad of H. bacteriophora migrates dorsally from the ventral vulval position and then migrates back around to the ventral position. The migrating arms cross each other near the vulva and extend beyond the vulva on either side. At around 48 h, the extending arms of the gonad of a young adult, grown from IJ, are visible near the vulva. Microinjection is performed at this position. Scale bar: 0.1 mm. Please click here to view a larger version of this figure.
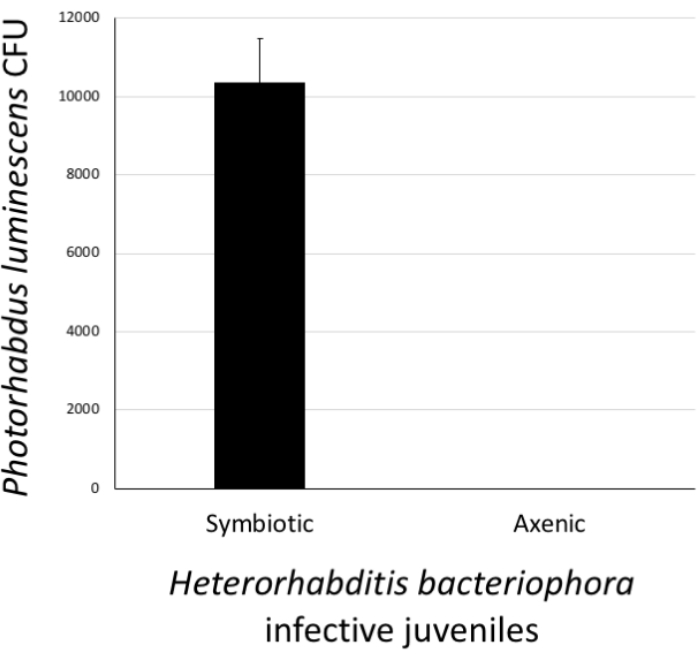
Figure 6: Validation of Heterorhabditis bacteriophora axenization. The success of the axenization procedure was estimated by confirming the absence of Photorhabdus luminescens bacterial cells in treated H. bacteriophora infective juveniles. For this, a pellet of approximately 500 surface-sterilized worms from the stock culture (symbiotic) or worms which had undergone the axenization process (axenic) were homogenized. Then, the homogenate was spread onto agar plates and following a 24 h incubation, the appearance of bacterial colonies (colony forming units, CFU) was monitored. Lack of bacterial colonies on the plates suggests that H. bacteriophora nematodes are free of their P. luminescens symbiotic bacteria. Please click here to view a larger version of this figure.
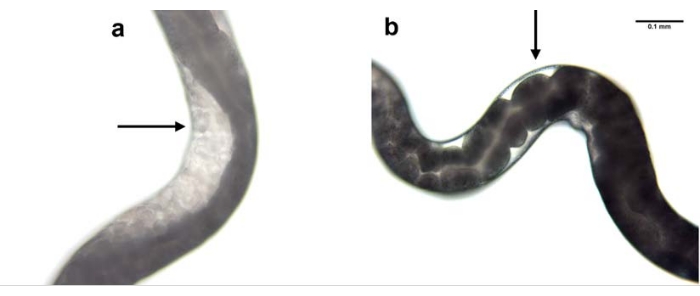
Figure 7: RNAi mediated knockdown of nol-5 gene. RNAi mediated knockdown of H. bacteriophora nol-5 gene results in a phenotype with no germline. The progeny of a wild type, non-injected H. bacteriophora nematode (a) contains eggs in its gonads. The progeny of a wild type, nol-5 RNAi injected H. bacteriophora nematode (b) contains no eggs in its empty gonads due to the knockdown of the nol-5 RNA. Scale bar: 0.1 mm. Please click here to view a larger version of this figure.
Discussion
Understanding the molecular basis of entomopathogenic nematode infection and insect anti-nematode immunity requires the separation of the parasites from the mutualistically associated bacteria13,15,16. The entomopathogenic nematodes H. bacteriophora and S. carpocapsae live together with the Gram-negative bacteria P. luminescens and X. nematophila, respectively17. Both bacterial species have been shown previously to encode factors that confer pathogenicity by targeting insect tissues and counteracting the innate immune system during infection18,19. This hinders efforts to identify the strategies that nematodes have evolved to interact with the insect host. Therefore, identification and functional characterization through gene knockdown of the nematode components that also participate in the infection process can be facilitated by the generation of axenic worms. Here, efficient protocols for producing axenic entomopathogenic nematodes and performing RNAi gene silencing in H. bacteriophora are presented.
A critical step in both protocols for successfully generating axenic nematodes is the surface-sterilization of H. bacteriophora and S. carpocapsae IJs14,20. This part of the method involves treatment of the worms with bleach solution and is considered crucial for the success of the axenization process because it removes the P. luminescens and X. nematophila bacteria from the nematode cuticle. This is an important procedure to ensure that only the mutualistic bacteria on the surface of the worms are cleared and not those inside the parasites. Completion of this step requires attention because extended bleach treatment will impact nematode survival.
A similarity of the present axenization protocol for S. carpocapsae nematodes with a previously established method is the use of the X. nematophila ΔrpoS mutant bacteria which are not able to colonize the worms21. A difference between the current methodology and a previously reported protocol, which introduced surface sterilized nematode eggs onto nutrient agar22, is the addition of antibiotics into the culture media to inhibit microbial contamination that would likely affect nematode growth and fitness.
RNAi by microinjection is an easy and reliable method of delivering the RNA to the oocytes. The burst of liquid inside the gonad provides visual confirmation of the release of the nucleic acid solution during the injection process. It was shown that RNAi by microinjection is significantly better than RNAi by soaking. This is probably due to the ability of the RNAi solution to reach oocytes that are at different stages of differentiation inside the gonad.
While optimizing the process, it is recommended to test different concentrations of RNA solution for better results. Various concentrations of RNA ranging from 50 ng/µL to 10 µg/µL were tested. It was found that a concentration of 6 µg/µL worked better than other concentrations. To increase the RNA yield during the in vitro transcription step, the protocol was modified from a 4 hour incubation period to 16 hours. This resulted in a substantial increase in the final yield of RNA. One of the key factors for successful microinjection is the amount of time that a nematode spends on the injection pad. Less time results in a healthy surviving nematode and healthy progeny with an increased chance of observing the intended phenotype.
The current protocol for culturing and genetically manipulating entomopathogenic nematodes is a significant contribution for future studies in the fields of nematology, immunology, and host-parasite interactions. Combining approaches that allow the manipulation of entomopathogenic nematodes will lead to the discovery of the genetic factors that define the interplay between nematode effector molecules and host immune signaling components that encode factors with anti-nematode properties. It will further determine which key nematode molecular components determine the symbiotic relationship with the related bacteria. Answering these questions is important for improving agricultural practices and developing novel means for the control of human parasitic nematodes.
Declarações
The authors have nothing to disclose.
Acknowledgements
We thank members of the Department of Biological Sciences at George Washington University for critical reading of the manuscript. All graphical figures were made using BioRender. Research in the I. E., J. H., and D. O'H. laboratories have been supported by George Washington University and Columbian College of Arts and Sciences facilitating funds and Cross-Disciplinary Research Funds.
Materials
| Agarose | VWR | 97062-244 | |
| Ambion Megascript T7 Kit | Thermo Fisher Scientific | AM1333 | |
| Ampicillin | Fisher Scientific | 611770250 | |
| Cell culture flask T25 | Fisher Scientific | 156367 | |
| Cell culture flask T75 | Fisher Scientific | 156499 | |
| ChoiceTaq Mastermix | Denville Scientific | C775Y42 | |
| Corn oil | VWR | 470200-112 | |
| Corn syrup | MP Biomedicals/VWR | IC10141301 | |
| Culture tube 10 mL | Fisher Scientific | 14-959-14 | |
| Eppendorf Femtotips Microloader Tips | Eppendorf | E5242956003 | |
| Ethanol | Millipore-Sigma | E7023 | |
| Falcon tube 50 mL | Fisher Scientific | 14-432-22 | |
| Femtojet Microinjector | Eppendorf | 5252000021 | |
| Filter paper | VWR | 28320-100 | |
| Galleria mellonella waxorms | Petco | – | |
| Glass coverslip | Fisher Scientific | 12-553-464 | 50 x 24 mm |
| Halocarbon Oil 700 | Sigma | H8898 | |
| Inoculating loop | VWR | 12000-806 | |
| Kanamycin | VWR | 97062-956 | |
| Kwik-Fil Borosilicate Glass Capillaries | World Precision Instruments | 1B100F-3 | 1.0 mm |
| LB Agar | Fisher Scientific | BP1425-500 | LB agar miller powder 500 g |
| LB Broth | Fisher Scientific | BP1426-500 | LB broth miller powder 500 g |
| Leica DM IRB Inverted Research Microscope | Microscope Central | – | |
| MacConkey medium | Millipore-Sigma | M7408-250G | |
| MEGAclear Transcription Clean-Up Kit | Thermo Fisher Scientific | AM1908 | |
| Microcentrifuge tube | VWR | 76332-064 | 1.5 ml |
| NanoDrop 2000 Spectrophotometer | Thermo Fisher Scientific | ND-2000 | |
| Needle syringe | VWR | BD305155 | 22G |
| Nutrient broth | Millipore-Sigma | 70122-100G | |
| Parafilm | VWR | 52858-076 | |
| Partitioned Petri dish | VWR | 490005-212 | |
| PBS | VWR | 97062-732 | Buffer PBS tablets biotech grade 200 tab |
| PCR primers | Azenta | – | |
| Pestle | Millipore-Sigma | BAF199230001 | Bel-Art Disposable Pestle |
| Petri dish 6 cm | VWR | 25384-092 | 60 x 15 mm |
| Petri dish 10 mm | VWR | 10799-192 | 35 x 10 mm |
| Proteose Peptone #3 | Thermo Fisher Scientific | 211693 | |
| Yeast extract | Millipore-Sigma | Y1625 |
Referências
- Lacey, L. A., et al. Insect pathogens as biological control agents: Back to the future. Journal of Invertebrate Pathology. 132, 1-41 (2015).
- Ozakman, Y., Eleftherianos, I. Nematode infection and antinematode immunity in Drosophila. Trends in Parasitology. 37 (11), 1002-1013 (2021).
- Kenney, E., Hawdon, J. M., O’Halloran, D. M., Eleftherianos, I. Secreted virulence factors from Heterorhabditis bacteriophora highlight its utility as a model parasite among Clade V nematodes. International Journal of Parasitology. 51 (5), 321-325 (2021).
- Bobardt, S. D., Dillman, A. R., Nair, M. G. The two faces of nematode infection: Virulence and immunomodulatory molecules from nematode parasites of mammals, insects and plants. Frontiers in Microbiology. 11, 2983 (2020).
- Castillo, J. C., Reynolds, S. E., Eleftherianos, I. Insect immune responses to nematode parasites. Trends in Parasitology. 27 (12), 537-547 (2011).
- Eleftherianos, I., Heryanto, C. Transcriptomic insights into the insect immune response to nematode infection. Genes. 12 (2), 202 (2021).
- Ciche, T. The biology and genome of Heterorhabditis bacteriophora. WormBook. , 1-9 (2007).
- Stock, S. P. Partners in crime: symbiont-assisted resource acquisition in Steinernema entomopathogenic nematodes. Current Opinion in Insect Science. 32, 22-27 (2019).
- Cao, M., Schwartz, H. T., Tan, C. -. H., Sternberg, P. W. The entomopathogenic nematode Steinernema hermaphroditum is a self-fertilizing hermaphrodite and a genetically tractable system for the study of parasitic and mutualistic symbiosis. Genética. 220 (1), (2021).
- Goodrich-Blair, H., Clarke, D. J. Mutualism and pathogenesis in Xenorhabdus and Photorhabdus: two roads to the same destination. Molecular Microbiology. 64 (2), 260-268 (2007).
- Abd-Elgawad, M. M. M. Photorhabdus spp.: An overview of the beneficial aspects of mutualistic bacteria of insecticidal nematodes. Plants. 10 (8), 1660 (2021).
- Dreyer, J., Malan, A. P., Dicks, L. M. T. Bacteria of the genus Xenorhabdus, a novel source of bioactive compounds. Frontiers in Microbiology. 9, 3177 (2018).
- Hallem, E. A., Rengarajan, M., Ciche, T. A., Sternberg, P. W. Nematodes, bacteria, and flies: a tripartite model for nematode parasitism. Current Biology. 17 (10), 898-904 (2007).
- Castillo, J. C., Shokal, U., Eleftherianos, I. A novel method for infecting Drosophila adult flies with insect pathogenic nematodes. Virulence. 3 (3), 339-347 (2012).
- Castillo, J. C., Shokal, U., Eleftherianos, I. Immune gene transcription in Drosophila adult flies infected by entomopathogenic nematodes and their mutualistic bacteria. Journal of Insect Physiology. 59 (2), 179-185 (2013).
- Eleftherianos, I., Joyce, S., Ffrench-Constant, R. H., Clarke, D. J., Reynolds, S. E. Probing the tri-trophic interaction between insects, nematodes and Photorhabdus. Parasitology. 137 (11), 1695-1706 (2010).
- Nielsen-LeRoux, C., Gaudriault, S., Ramarao, N., Lerelcus, D., Givaudan, A. How the insect pathogen bacteria Bacillus thuringiensis and Xenorhabdus/Photorhabdus occupy their hosts. Current Opinion in Microbiology. 15 (3), 220-231 (2012).
- Waterfield, N. R., Ciche, T., Clarke, D. Photorhabdus and a host of hosts. Annual Review of Microbiology. 63, 557-574 (2009).
- Ozakman, Y., Eleftherianos, I. Immune interactions between Drosophila and the pathogen Xenorhabdus. Microbiological Research. 240, 126568 (2020).
- Yadav, S., Shokal, U., Forst, S., Eleftherianos, I. An improved method for generating axenic entomopathogenic nematodes. BMC Research Notes. 8 (1), 1-6 (2015).
- Mitani, D. K., Kaya, H. K., Goodrich-Blair, H. Comparative study of the entomopathogenic nematode, Steinernema carpocapsae, reared on mutant and wild-type Xenorhabdus nematophila. Biological Control. 29 (3), 382-391 (2004).
- McMullen, J. G., Stock, S. P. In vivo and in vitro rearing of entomopathogenic nematodes (Steinernematidae and Heterorhabditidae). Journal of Visualized Experiments. (91), e52096 (2014).
