Fånga kromosomkonformation över längdskalor
Summary
Hi-C 3.0 är ett förbättrat Hi-C-protokoll som kombinerar formaldehyd- och disuccinimidylglutarat-tvärbindare med en cocktail av DpnII- och DdeI-restriktionsenzymer för att öka signal-brusförhållandet och upplösningen av kromatininteraktionsdetektering.
Abstract
Kromosomkonformationsfångst (3C) används för att detektera tredimensionella kromatininteraktioner. Vanligtvis används kemisk tvärbindning med formaldehyd (FA) för att fixa kromatininteraktioner. Därefter omvandlar kromatinsmältning med ett restriktionsenzym och efterföljande religering av fragmentändar tredimensionell (3D) närhet till unika ligeringsprodukter. Slutligen, efter omkastning av tvärbindningar, proteinavlägsnande och DNA-isolering, skjuvas DNA och förbereds för sekvensering med hög genomströmning. Frekvensen av närhetsligering av par av loci är ett mått på frekvensen av deras samlokalisering i tredimensionellt utrymme i en cellpopulation.
Ett sekvenserat Hi-C-bibliotek ger genomomfattande information om interaktionsfrekvenser mellan alla par av loci. Upplösningen och precisionen hos Hi-C är beroende av effektiv tvärbindning som upprätthåller kromatinkontakter och frekvent och enhetlig fragmentering av kromatinet. Detta dokument beskriver ett förbättrat Hi-C-protokoll på plats , Hi-C 3.0, som ökar effektiviteten för tvärbindning genom att kombinera två tvärbindare (formaldehyd [FA] och disuccinimidylglutarat [DSG]), följt av finare matsmältning med två restriktionsenzymer (DpnII och DdeI). Hi-C 3.0 är ett enda protokoll för exakt kvantifiering av genomvikningsfunktioner i mindre skalor som slingor och topologiskt associerande domäner (TADs), liksom funktioner i större kärnomfattande skalor som fack.
Introduction
Kromosomkonformationsfångst har använts sedan 20021. I grund och botten är varje konformationsfångstvariant beroende av fixering av DNA-protein och protein-proteininteraktioner för att bevara 3D-kromatinorganisation. Detta följs av DNA-fragmentering, vanligtvis genom restriktionssmältning, och slutligen religation av närliggande DNA-ändar för att omvandla rumsligt proximala loci till unika kovalenta DNA-sekvenser. Initiala 3C-protokoll använde PCR för att prova specifika “en-till-en” -interaktioner. Efterföljande 4C-analyser möjliggjorde detektering av “en-till-alla” interaktioner2, medan 5C detekterade “många-till-många” interaktioner3. Kromosomkonformationsfångst kom till full frukt efter att ha implementerat nästa generations sekvensering med hög genomströmning (NGS), vilket möjliggjorde detektion av “allt-till-alla” genomiska interaktioner med hjälp av genomomfattandeHi-C4 och jämförbara tekniker som 3C-seq5, TCC6 och Micro-C 7,8 (se även granskning av Denker och De Laat9).
I Hi-C används biotinylerade nukleotider för att markera 5 ′ överhäng efter matsmältning och före ligering (figur 1). Detta möjliggör val av korrekt smälta och religerade fragment med hjälp av streptavidinbelagda pärlor, vilket skiljer det från GCC10. En viktig uppdatering av Hi-C-protokollet implementerades av Rao et al.11, som utförde matsmältningen och religationen i intakta kärnor (dvs. in situ) för att minska falska ligeringsprodukter. Dessutom minskade fragmentstorleken och ökade upplösningspotentialen för Hi-rötning genom att ersätta HindIII-matsmältningen med MboI (eller DpnII). Denna ökning möjliggjorde detektion av relativt småskaliga strukturer och en mer exakt genomisk lokalisering av kontaktpunkter, såsom DNA-slingor mellan små cis-element, t.ex. slingor mellan CTCF-bundna platser genererade av loopextrudering11,12. Denna potential kostar dock. För det första kräver en tvåfaldig ökning av upplösningen en fyrfaldig (22) ökning av sekvenseringsläsningarna13. För det andra ökar de små fragmentstorlekarna möjligheten att missta osmälta angränsande fragment för smälta och religerade fragment14. Som nämnts skiljer sig i Hi-C smälta och religerade fragment från osmälta fragment genom närvaron av biotin vid ligeringskorsningen. Korrekt borttagning av biotin från oligierade ändar krävs dock för att säkerställa att endast ligeringskorsningar dras ner14,15.
Med den minskande kostnaden för NGS blir det möjligt att studera kromosomveckning mer detaljerat. För att minska storleken på DNA-fragment och därmed öka upplösningen kan Hi-C-protokollet anpassas för att oftare använda skärande restriktionsenzymer16 eller för att använda kombinationer av restriktionsenzymer17,18,19. Alternativt kan MNase 7,8 i Micro-C och DNase i DNase Hi-C20 titreras för att uppnå optimal matsmältning.
En nyligen genomförd systematisk utvärdering av grunderna i 3C-metoder visade att detekteringen av kromosomvikningsfunktioner på varje längdskala förbättrades avsevärt med sekventiell tvärbindning med 1% FA följt av 3 mM DSG17. Dessutom var Hi-C med HindIII-matsmältning det bästa alternativet för att upptäcka storskaliga vikningsfunktioner, såsom fack, och att Micro-C var överlägsen för att upptäcka småskaliga vikningsfunktioner som DNA-slingor. Dessa resultat ledde till utvecklingen av en enda, högupplöst “Hi-C 3.0” -strategi, som använder kombinationen av FA- och DSG-tvärbindare följt av dubbel matsmältning med DpnII- och DdeI-endonukleaser21. Hi-C 3.0 ger en effektiv strategi för allmän användning eftersom den exakt upptäcker vikningsfunktioner över alla längdskalor17. Den experimentella delen av Hi-C 3.0-protokollet beskrivs här och typiska resultat som kan förväntas efter sekvensering visas.
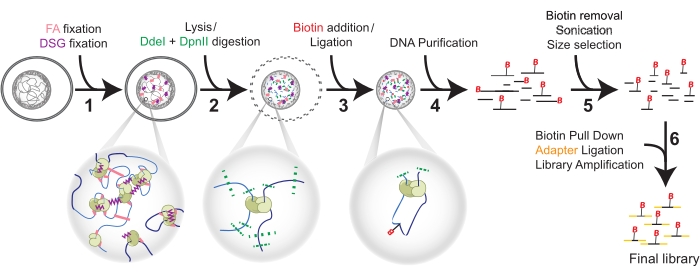
Bild 1: Hi-C-procedur i sex steg. Celler fixeras först med FA och sedan DSG (1). Därefter föregår lys en dubbel matsmältning med DdeI och DpnII (2). Biotin tillsätts genom överhängsfyllning och proximala trubbiga ändar ligeras (3) före DNA-rening (4). Biotin avlägsnas från oligata ändar före ultraljudsbehandling och storleksval (5). Slutligen möjliggör neddragning av biotin adapterligering och biblioteksförstärkning med PCR (6). Förkortningar: FA = formaldehyd; DSG = disuccinimidylglutarat; B = Biotin. Klicka här för att se en större version av denna siffra.
Protocol
Representative Results
Discussion
Kritiska steg för cellhantering
Även om det är möjligt att använda ett lägre antal ingångsceller har detta protokoll optimerats för ~ 5 × 106 celler per sekvenseringsfält (~ 400 M läsningar) för att säkerställa korrekt komplexitet efter djup sekvensering. Celler räknas bäst före fixering. För generering av ultradeep-bibliotek multiplicerar vi i allmänhet antalet banor (och celler) tills önskat läsdjup har uppnåtts. För optimal fixering bör seruminnehållande medium ersättas med PBS före FA-fixering, och fixeringslösningar bör tillsättas omedelbart och utan koncentrationsgradienter15,22. För cellskörd föredras skrapning framför trypsinisering, eftersom övergången från en plattare till en sfärisk form efter trypsinisering kan påverka kärnkonformationen. Efter tillsats av DSG förloras lätt lösa och klumpiga cellpellets. Var försiktig när du hanterar celler i detta skede och lägg till upp till 0.05% BSA för att minska klumpningen.
Ändringar av metoden
Detta protokoll utvecklades med hjälp av mänskliga celler17. Ändå, baserat på erfarenhet av kromosomkonformationsfångst, bör detta protokoll fungera för de flesta eukaryota celler. För en betydligt lägre ingång (~ 1 × 106 celler) rekommenderar vi att du använder hälften av volymerna för lys- och konformationsupptagningsprocedurerna [steg 2.1-2.4]. Detta skulle också göra det möjligt att utföra DNA-isolering [steg 2.5] i en bordscentrifug med 1,7 ml rör, vilket kan förbättra pelleteringen för låga DNA-koncentrationer. Kvantifieringen av DNA (steg 2.6.6) kommer att indikera hur man ska gå vidare. För låga mängder isolerat DNA (1-5 μg) föreslår vi att du hoppar över storleksvalet (steg 3.3) och fortsätter med biotinborttagning efter att ha minskat volymen från 130 μL till ~ 45 μL med en CFU.
Detta protokoll utvecklades specifikt för att säkerställa högkvalitativa data efter efterföljande tvärbindning med FA och DSG och matsmältning med DpnII och DdeI. Alternativa tvärbindningsstrategier som FA följt av EGS (etylenglykolbis(succinimidylsuccinat)), som också används i ChIP-seq23 och ChIA-PET24, kan dock fungera lika bra17. På liknande sätt kan olika enzymkombinationer, såsom DpnII och HinfI18 eller MboI, MseI och NlaIII19 användas för matsmältning. När du anpassar enzymkombinationer, var noga med att använda biotinylerade nukleotider som kan fylla i de specifika 5′-överhängen och använda de mest optimala buffertarna för varje cocktail. DpnII levereras med sin egen buffert och enzymtillverkaren rekommenderar en specifik buffert för DdeI-matsmältning. Men för dubbel matsmältning med DpnII och DdeI i detta protokoll rekommenderas begränsningsbuffert eftersom den är klassad till 100% aktivitet för båda enzymerna.
Felsöka konformationsavskiljning
De tre viktigaste stegen i kromosomkonformationsfångst: tvärbindning, matsmältning och relikatering har alla utförts innan resultaten kan visualiseras på gel. För att bestämma kvaliteten på vart och ett av dessa tre steg och urskilja var problem kan ha uppstått tas alikvoter före (CI) och efter matsmältningen (DC) och laddas på gelén tillsammans med det ligerade Hi-C-provet (figur 2). Denna gel används för att bestämma kvaliteten på Hi-C-provet och om det kommer att vara värt att fortsätta protokollet. Utan CI och DC är det svårt att hitta potentiella suboptimala steg. Det är värt att notera att suboptimal ligering kan bero på ett problem i själva ligeringen, fyllningen eller ett problem med tvärbindning. För att felsöka tvärbindning, se till att inte använda mer än 1 × 107 celler per bibliotek och börja med färska tvärbindningsreagenser och rena celler (dvs. sköljda med PBS). För ligering, se till att celler och ligeringsblandning hålls på is. Tillsätt T4 DNA-ligas strax före 4-timmarsinkubationen vid 16 °C och blanda väl.
Felsöka biblioteksförberedelser
Om mer än 10 PCR-cykler behövs eller ingen PCR-produkt kan ses på gel efter PCR-titrering (figur 4), finns det några alternativ för att spara Hi-C-provet. När du arbetar tillbaka från PCR-titreringen är det första alternativet att prova PCR igen. Om det fortfarande inte finns tillräckligt med produkt är det möjligt att prova ytterligare en omgång A-tailing och adapter ligering (steg 3.6) efter att ha tvättat pärlorna två gånger med 1x TLE-buffert. Efter denna ytterligare A-tailing och adapter ligering kan man fortsätta till PCR-titreringen som tidigare. Om det fortfarande inte finns någon produkt är det sista alternativet att resonikera 0,8x-fraktionen från steg 3.3 och fortsätta därifrån.
Begränsningar och fördelar med Hi-C3.0
Det är viktigt att inse att Hi-C är en populationsbaserad metod som fångar den genomsnittliga frekvensen av interaktioner mellan par av loci i cellpopulationen. Vissa beräkningsanalyser är utformade för att skilja kombinationer av konformationer från en population25, men i princip är Hi-C blind för skillnader mellan celler. Även om det är möjligt att utföra encells Hi-C26,27 och beräkningsinferenser kan göras28, är encells Hi-C inte lämplig för att erhålla 3C-information med ultrahög upplösning. En ytterligare begränsning av Hi-C är att den bara upptäcker parvisa interaktioner. För att upptäcka multikontaktinteraktioner kan man antingen använda frekventa skärare i kombination med kortläsningssekvensering (Illumina)16 eller utföra multikontakt 3C29 eller 4C30, med hjälp av långläsningssekvensering från PacBio- eller Oxford Nanopore-plattformar. Hi-C-derivat för att specifikt detektera kontakter mellan och längs systerkromatider har också utvecklats31,32.
Även om Hi-C19 och Micro-C33 kan användas för att generera kontaktkartor vid subkilobasupplösningar, kräver båda en stor mängd sekvensläsningar och detta kan bli ett kostsamt företag. För att komma till liknande eller ännu högre upplösning utan kostnader kan anrikning för specifika genomiska regioner (capture-C 34) eller specifika proteininteraktioner (ChiA-PET 35, PLAC-seq36, Hi-ChIP37) tillämpas. Styrkan och nackdelen med dessa berikningsapplikationer är att endast ett begränsat antal interaktioner samplas. Med sådana anrikningar förloras den globala aspekten av Hi-C (och möjligheten till global normalisering).
Betydelsen och potentiella tillämpningar av Hi-C3.0
Detta protokoll har utformats för att möjliggöra högupplöst, ultradeep 3C samtidigt som det upptäcker storskaliga vikningsfunktioner som TADs och fack17 (figur 6). Detta protokoll börjar med 5 × 106 celler per rör för varje Hi-C-bibliotek, vilket bör vara mer än tillräckligt med material för att sekvensera en eller två banor på en flödescell för att få upp till 1 miljard parade slutläsningar. För ultradeep-sekvensering bör flera rör med 5 × 106 celler förberedas, beroende på antalet mappade läsningar och PCR-dubbletter. Vid den högsta upplösningen (<1 kb) finns looping-interaktioner mestadels mellan CTCF-webbplatser, men promotor-förstärkarinteraktioner kan också detekteras. Läsare kan hänvisa till Akgol Oksuz et al.17 för en detaljerad beskrivning av dataanalysen.
Declarações
The authors have nothing to disclose.
Acknowledgements
Vi vill tacka Denis Lafontaine för protokollutveckling och Sergey Venev för bioinformatisk hjälp. Detta arbete stöddes av ett bidrag från National Institutes of Health Common Fund 4D Nucleome Program till JD (U54-DK107980, UM1-HG011536). JD är utredare vid Howard Hughes Medical Institute.
Denna artikel lyder under HHMI:s policy för öppen tillgång till publikationer. HHMI-labbchefer har tidigare beviljat en icke-exklusiv CC BY 4.0-licens till allmänheten och en underlicensierbar licens till HHMI i sina forskningsartiklar. I enlighet med dessa licenser kan det författargodkända manuskriptet till denna artikel göras fritt tillgängligt under en CC BY 4.0-licens omedelbart efter publicering.
Materials
| 1 kb Ladder | New England Biolabs | N3232L | |
| Agarose | Invitrogen | 16500100 | |
| Agencourt AMPure XP magnetic beads , 60 mL | Beckman Coulter | A63881 | |
| Amicon Ultra-0.5 Centrifugal Filter Unit (CFU) | EMD Millipore | UFC500396 | |
| Annealing Buffer (5x) | See recipe in supplemental materials | ||
| ATP 10 mM | ThermoFisher | R0441 | |
| Avanti J-25i High Speed Refrigerated ultra-centrifuge | Beckman Coulter | ||
| beckman ultracentrifuge tube 35 mL | Beckman Coulter | 357002 | |
| Binding Buffer (2x) | See recipe in supplemental materials | ||
| biotin-14-dATP 0.4 mM | Invitrogen | 19524-016 | |
| BSA 10 mg/mL | New England Biolabs | B9000S | dilute from 20 mg/mL |
| Cell scraper | Falcon | 353089 | |
| Cell scraper | Corning | 3008 | |
| Conical polypropylene tubes 50 mL | Denville | C1062-P | |
| Conical tube 15 mL | Denville | C1017-P | |
| Covaris micro tube AFA fiber with snap-cap 130 µL | Covaris | 520045/520077 | |
| Covaris Sonicator | Covaris | E220/E220evolution/M220 | |
| Culture flask 175 cm2 | Falcon | 353112 | |
| Culture plates 150 mm x 25 mm | Corning | 430599 | |
| dATP 1 mM | Invitrogen | 56172 | |
| dATP 10 mM | Invitrogen | 56172 | |
| dCTP 10 mM | Invitrogen | 56173 | |
| DdeI | New England Biolabs | R0175L | |
| dGTP 10 mM | Invitrogen | 56174 | |
| DMSO | Sigma | D2650-5x10ML | |
| dNTP mix 25 mM | Invitrogen | 10297117 | |
| Dounce homogenizer | DWK Life Sciences | 8853010002/8853030002 | |
| DPBS | Gibco | 14190-144 | |
| DpnII | New England Biolabs | R0543M | |
| DSG | ThermoScientific | 20593 | |
| dTTP 10 mM | Invitrogen | 56175 | |
| Ethanol 70% | Fisher | A409-4 | Diluted from 100% |
| Ethidium Bromide | Fisher | BP1302-10 | |
| Formaldehyde (37%) | Fisher | BP531-500 | |
| Gel loading dye (6x ) | New England Biolabs | B7024S | |
| Glycine in ultrapure water 2.5 M | Sigma | G8898-1KG | |
| HBSS | Gibco | 14025-092 | |
| Igepal CA-630 detergent | MP Biomedicals | 198596 | |
| Klenow DNA polymerase 5 U/µL | New England Biolabs | M0210L | |
| Klenow Fragment 3–>5’ exo-, 5 U/µL | New England Biolabs | M0212L | |
| ligation buffer (10x) | New England Biolabs | B7203S | |
| Liquid nitrogen | |||
| LoBind microcentrifuge tube 1.7 mL | Eppendorf | 22431021 | |
| Low Molecular Weight DNA Ladder | New England Biolabs | N3233L | |
| Lysis buffer | See recipe in supplemental materials | ||
| Magnetic Particle separator | ThermoFisher | 12321D | |
| Microfuge tubes 1.7 mL | Axygen | MCT-175-C | |
| MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| NEBuffer 2.1 (10x) | New England Biolabs | B7002S | |
| NEBuffer 3.1 (10x) | New England Biolabs | B7203S | |
| PBS | Gibco | 70013-032 | |
| PCR (strip) tubes | Biorad | TBS0201/ TCS0803 | |
| PCR thermocycler | Biorad | T100 | |
| Pfu Ultra II Buffer (10x) | Agilent | Comes with Pfu Ultra | |
| PfuUltra II Fusion HS DNA Polymerase | Agilent | 600674 | |
| Phase lock tube 15 mL | Qiagen | 129065 | |
| Phase lock tubes 2 mL | Qiagen | 129056 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593-049 | |
| Protease inhibitor cocktail | ThermoFisher | 78440 | |
| Proteinase K in ultrapure water 10 mg/mL | Invitrogen | 25530-031 | |
| Refrigerated Centrifuge | Eppendorf | 5810R | |
| RNase A, DNase and protease-free 10 mg/mL | Thermo Scientific | EN0531 | |
| Rotator | Argos technologies | EW-04397-40 or rocking platform | |
| SDS 1% | Fisher | BP13111 | |
| Sodium acetate pH = 5.2, 3 M | Sigma | ||
| Sub-Cell GT Horizontal Electrophoresis System | Biorad | 1704401 | |
| T4 DNA ligase 1 U/µL | Invitrogen | 100004817 | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase 3 U/µL | New England Biolabs | M0203L | |
| T4 ligation buffer (5x) | Invitrogen | Y90001 | |
| T4 polynucleotide kinase 10 U/µL | New England Biolabs | M0201L | |
| Tabletop centrifuge | Eppendorf | 5425 | |
| TBE buffer | See recipe in supplemental materials | ||
| Tris Low EDTA Buffer (TLE) | See recipe in supplemental materials | ||
| Triton X-100 (10%) | Sigma | 93443 | |
| Truseq adapter oligos | Integrated DNA Technologies (IDT)) | https://www.idtdna.com/site/order/oligoentry | 250 nmole and HPLC purified |
| Tween 20 detergent | Fisher | 9005-64-5 | |
| Tween Wash Buffer | See recipe in supplemental materials | ||
| Vortex | Scientific Industries | (G560)SI-0236 |
Referências
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Stadhouders, R., et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. 8 (3), 509-524 (2013).
- Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F., Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nature Biotechnology. 30 (1), 90-98 (2012).
- Hsieh, T. H., et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 162 (1), 108-119 (2015).
- Hsieh, T. -. H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Rodley, C. D., Bertels, F., Jones, B., O’Sullivan, J. M. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genetics and Biology. 46 (11), 879-886 (2009).
- Rao, S. S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Alipour, E., Marko, J. F. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research. 40 (22), 11202-11212 (2012).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- Golloshi, R., Sanders, J. T., McCord, R. P. Iteratively improving Hi-C experiments one step at a time. Methods. 142, 47-58 (2018).
- Darrow, E. M., et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America. 113 (31), 4504-4512 (2016).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Ghuryeid, J., et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Computational Biology. 15 (8), 1007273 (2019).
- Gu, H., et al. Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. , (2021).
- Ramani, V., et al. Mapping 3D genome architecture through in situ DNase Hi-C. Nature Protocols. 11 (11), 2104-2121 (2016).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. Current Protocols. 1 (7), 198 (2021).
- Belton, J. -. M. M., et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58 (3), 268-276 (2012).
- Truch, J., Telenius, J., Higgs, D. R., Gibbons, R. J. How to tackle challenging ChIP-Seq, with long-range cross-linking, Using ATRX as an example. Methods in Molecular Biology. 1832, 105-130 (2018).
- Wang, P., et al. In situ chromatin interaction analysis using paired-end tag sequencing. Current Protocols. 1 (8), 174 (2021).
- Tjong, H., et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proceedings of the National Academy of Sciences of the United States of America. 113 (12), 1663-1672 (2016).
- Nagano, T., et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 547 (7661), 61-67 (2017).
- Ramani, V., et al. Massively multiplex single-cell Hi-C. Nature Methods. 14 (3), 263-266 (2017).
- Meng, L., Wang, C., Shi, Y., Luo, Q. Si-C is a method for inferring super-resolution intact genome structure from single-cell Hi-C data. Nature Communications. 12 (1), 4369 (2021).
- Tavares-Cadete, F., Norouzi, D., Dekker, B., Liu, Y., Dekker, J. Multi-contact 3C reveals that the human genome during interphase is largely not entangled. Nature Structural & Molecular Biology. 27 (12), 1105-1114 (2020).
- Vermeulen, C., et al. Multi-contact 4C: long-molecule sequencing of complex proximity ligation products to uncover local cooperative and competitive chromatin topologies. Nature Protocols. 15 (2), 364-397 (2020).
- Oomen, M. E., Hedger, A. K., Watts, J. K., Dekker, J. Detecting chromatin interactions between and along sister chromatids with SisterC. Nature Methods. 17 (10), 1002-1009 (2020).
- Mitter, M., et al. Conformation of sister chromatids in the replicated human genome. Nature. 586 (7827), 139-144 (2020).
- Krietenstein, N., et al. Ultrastructural Details of Mammalian Chromosome Architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462 (7269), 58-64 (2009).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
