Validation of single-cell collection is performed visually during LCM procedures. Cell nuclei are assessed at the QC station. The cell type can be determined by emission of tagged fluorophore for that cell type and its general morphology. If non-desired cells have been selected on the cap, their genetic material can be destroyed with a UV laser at the QC station. Further validation by molecular analysis is also necessary. In this representative example16, two types of neurons were selected-tyrosine hydroxylase (Th) positive (+) and Th negative (-), in addition to microglia. Figure 1D demonstrates that samples meant to be neurons demonstrated statistically significant elevated expression of the neuronal marker, NeuN. Concurrently, microglial samples had significant elevation of the microglial markers, CD34 and Cx3xr1, suggesting that sample selection was performed with high fidelity. Additionally, Th+ neuronal samples demonstrated significantly elevated expression of Th compared to Th- neuronal samples and microglia samples while Th- neurons demonstrated significantly elevated expression of GCG (a preproglucagon coding gene) suggesting that these Th- neuronal samples are enriched with neurons that use GLP-1 as a neurotransmitter (Figure 1D). A more global computational measure known as linear discriminate analysis showed that these three cell types had differing gene expression profiles across all 65 genes measured with neurons and microglia separating along the x-axis and Th+ and Th- neuronal samples dividing along the y-axis (Figure 1E).
The quality of the data was also validated with intra and inter-batch technical replicates (Figure 2 and Figure 3). Intra-batch replicates control for the quality of data from that specific batch. If intra-batch replicates do not align, another qPCR chip experiment can be performed from the same sample and assay plates that were made to run the first batch. Inter-batch replicates ensure that data from across batches can be compared. This is crucial for experiments in which large numbers of samples are assayed requiring multiple batches such as this representative example. There was one outlier in this experiment as sample 40 in batch 4 (Figure 3). However, sample 40 in batches 1, 2, and 3 correctly aligned, and the other replicates from batch 4 aligned with batches 1, 2, and 3 suggesting that this was an isolated bad reaction, which occurred in this one replicate. Comparing across batches also requires proper data normalization techniques. This experiment used median centering, though housekeeping genes (Actb, Gapdh, Ldha), were also assayed in this experiment and served as a control for the median-centering method. That is, both methods yielded highly analogous datasets further suggesting high data quality.
Figure 4 displays a heat map of GLP-1 enriched neuronal samples that is organized by cellular subphenotype. This figure not only demonstrates the importance of cellular subphenotypes in neurobiology, but also demonstrates how ratios of subphenotypes change through alcohol withdrawal. The heat map shows that subphenotype A, which highly expresses the inflammatory gene cluster 1, increases in ratio at the 8 h Wd time point and reaches a maximum at 32 h Wd. By 176 h Wd, this inflammatory subphenotype is normalizing its prevalence back to control. Subphenotype B, which highly expresses GABAR gene cluster 2, demonstrates an overall suppression in the expression of this gene cluster by the 176 h Wd condition. These findings demonstrate how this GLP-1 neuronal cell type becomes hyperexcitable by increasing its inflammatory subphenoype and concurrently decreasing the level of expression of GABARs in its GABA subphenotype during alcohol withdrawal. Figure 5 combines the gene expression of individual samples into averages so that the expression of clusters of genes and the location of the protein of that gene transcript can be visualized throughout the time series.
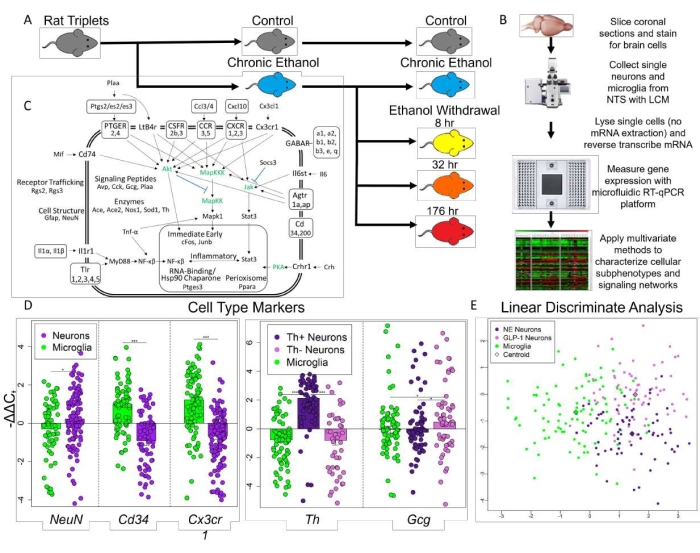
Figure 1: Experimental design and single-cell selection. (A) Rat triplets were randomly assigned to one of the five treatment conditions. (B) Single-cell transcriptome data generation. (C) Cartoon representation of genes assayed and their function. Genes in green were not assayed. Official gene symbol is used. (D) Gene expression of cell type markers. Error bars show standard error. Neurons compared to microglia, p-values = 0.0273, 3.94 x 10-10, 7.73 x 10-12, respectively. Th+ neurons showed elevated Th expression compared to Th- neurons (p = 4.56 x 10-11) and microglia (p = 2.95678 x 10-15). Th- neurons showed elevated expression of GCG compared to Th+ neurons (p = 0.0106) and microglia (p = 0.0435) indicating they are GLP-1+ neurons. *p < 0.05, ***p < 4 x 10-10. (E) Linear discriminate analysis of all samples displays the difference across all genes measured between the three cell types collected in a two-dimension space. Centroid distance between NE neurons and GLP-1 neurons = 3.30, NE neurons and microglia = 1.57, GLP-1 Neurons and microglia = 2.92. This figure has been modified from O'Sullivan et al. 202116. Please click here to view a larger version of this figure.
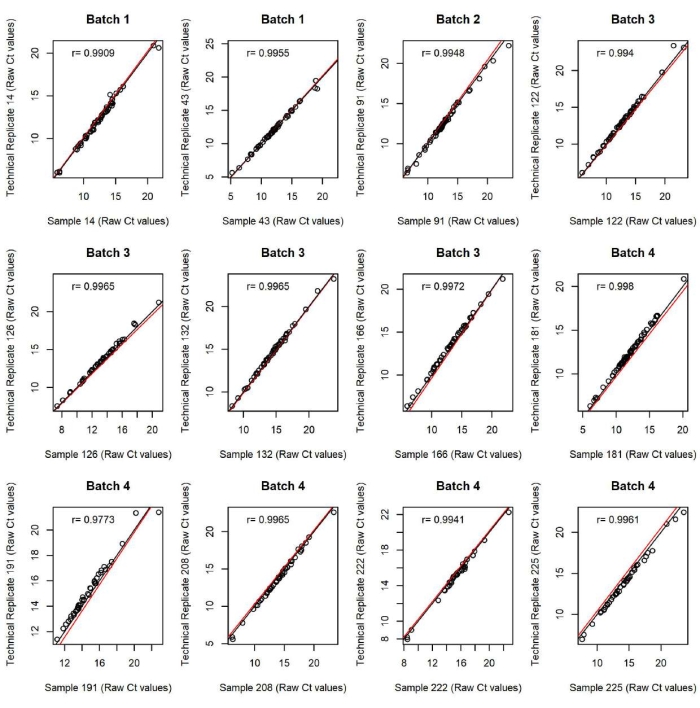
Figure 2: Technical replicate plots of raw Ct values within a batch. Each graph displays raw Ct values for intrachip technical replicates plotted against each other demonstrating technical experimental integrity. This figure has been modified from O'Sullivan et al. 202116. Please click here to view a larger version of this figure.
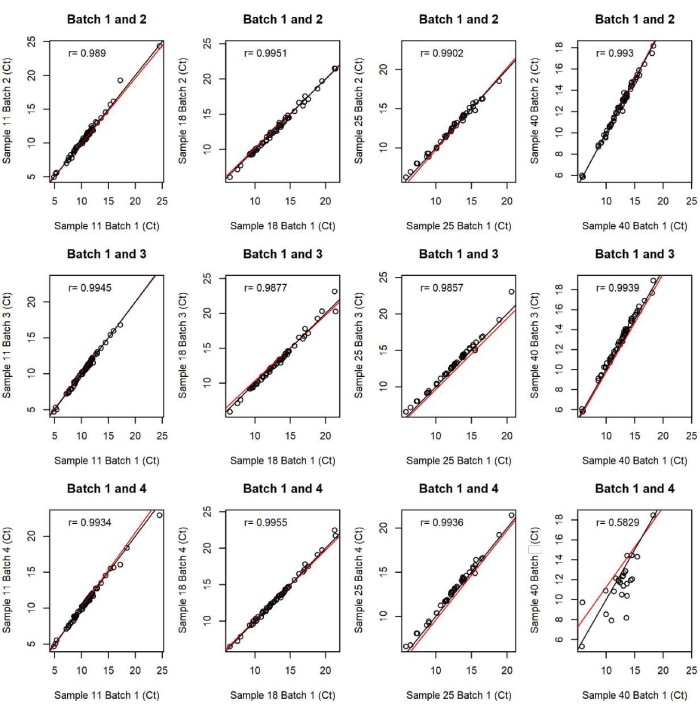
Figure 3: Technical replicate plots of raw Ct values between batches. Each graph displays raw Ct values for interchip technical replicates plotted against each other demonstrating that all batches are comparable to each other. This figure has been modified from O'Sullivan et al. 202116. Please click here to view a larger version of this figure.
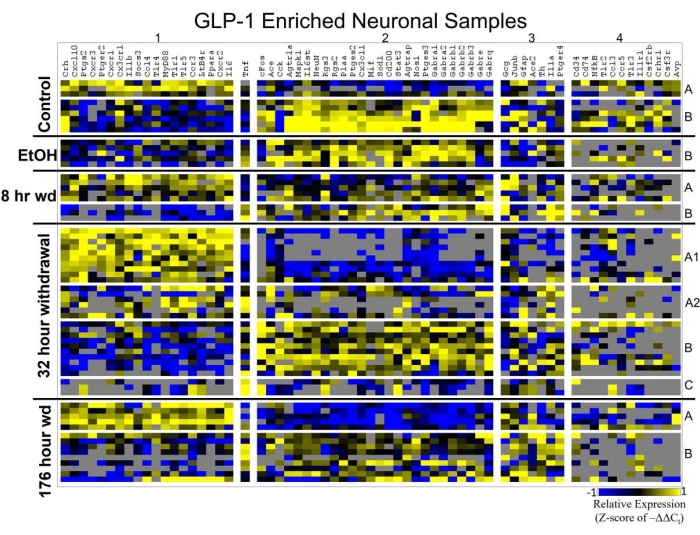
Figure 4: Heat map of GLP-1 enriched neuronal samples. Heat map displays cellular subphenotypes of GLP-1 enriched neuron samples through an alcohol withdrawal time series. Rows represent 10-cell pooled samples with cellular subphenotype clusters labeled with uppercase letters. Columns represent the z-score of -ΔΔCt gene expression values on a -1 to +1 color scale for that gene in that sample. Gene clusters are labeled by number. This figure has been modified from O'Sullivan et al. 202116. Please click here to view a larger version of this figure.
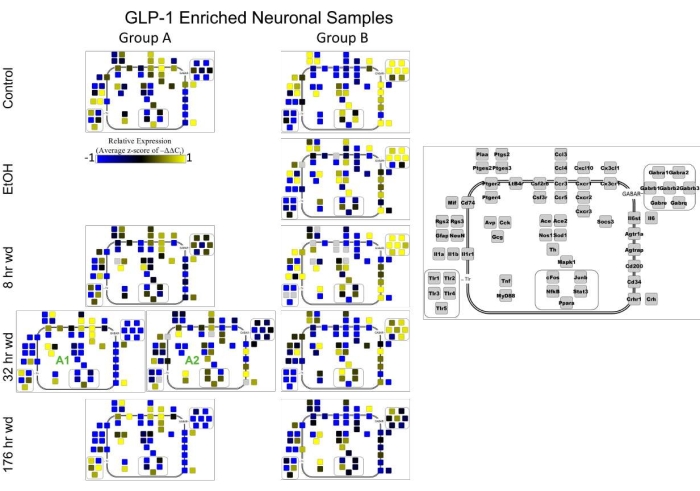
Figure 5: Suphenotype gene expression in GLP-1 enriched neuronal samples. Cellular diagrams display boxes representing relative gene expression (average z-score of -ΔΔCt values) of subphenotypes shown in the heatmap in Figure 4. The diagrams were constructed using Cytoscape version 3.8.0 and z-scores were calculated using the scale function in R version 3.5.2. -ΔΔCt labels on the right show which boxes correspond to which gene and the color represents expression (blue is low expression and yellow is high expression). The location of the box represents the localization or function of the protein product from that gene transcript. Green numbers indicate subgroups within subphenotypes. This figure has been modified from O'Sullivan et al. 202116. Please click here to view a larger version of this figure.
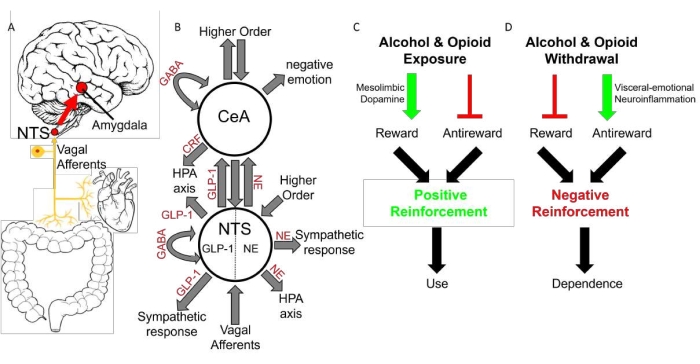
Figure 6: Interoceptive vagal circuit, visceral-emotional neuraxis, and schematic of opponent-process model of addiction. (A) Interoceptive vagal afferents relay the state of the gut, which is highly influenced by gut microflora, and other peripheral organs to the nucleus tractus solitarius (NTS). This information is subsequently relayed to the amygdala and influences emotional states. (B) A simplified cartoon representation showing the integrative roles of the nucleus tractus solitarius (NTS) and the central nucleus of the amygdala (CeA) in emotion, stress, and autonomic regulation. Two neuronal subtypes, GLP-1 and NE neurons are highlighted. Many anatomical and functional connections are omitted for clarity. Abbreviations: NE = norepinephrine; GLP-1 = glucagon-like peptide 1; GABA = γ-aminobutyric acid; HPA axis = hypothalamic-pituitary-adrenal axis; CRF = corticotropin releasing hormone. (C) Alcohol and/or opioid exposure has two actions: stimulate reward, via the mesolimbic dopamine pathway, and inhibit antireward. These actions motivate substance use via positive reinforcement. (D) Alcohol and/or opioid withdrawal has two actions: inhibit reward, by inhibiting the mesolimbic dopamine pathway (not shown) and stimulate antireward. This study proposes that visceral-emotional neuroinflammation is an endpoint in antireward stimulation, though this hypothesis warrants further testing. These actions, whatever the mechanism, motivate substance dependence via negative reinforcement. This figure has been modified from O'Sullivan et al. 202116. Please click here to view a larger version of this figure.
Supplementary Table 1: Primers for microfluidic RT-qPCR. All primer pairs for mRNA amplification are listed along with the amplicon length formed by each primer pair. This table has been modified from O'Sullivan et al. 202116. Please click here to download this File.
