The protocol describes a flow cytometric approach to identify BFU-es and CFU-es in freshly harvested bone marrow and spleen cells. It starts with harvesting fresh BM and spleen from mice and immediately placing the tissue on ice. All procedures are conducted in the cold to preserve cell viability. Cells are labeled with a "lineage" antibody cocktail that allows the exclusion of all cells expressing markers of differentiated blood lineages (the FITC- Lin cocktail, Table 3, in the case of flow cytometric analysis; or magnetic bead enrichment, Table 4, for lineage-negative cells). Cells are also labeled with antibodies against markers that allow us to distinguish a number of early progenitor populations within the lineage-negative cell fraction. Labeling is followed by either flow cytometric sorting or analysis. The approach to flow cytometric sorting is similar to that of flow cytometric analysis but is preceded by magnetic-bead enrichment for the Lin– cell fraction to reduce the time and expense of sorting large numbers of cells. When sorting progenitors, BM is pooled from multiple mice, the number depending on the downstream application. Typically, BM from a total of 10 mice (approximately 1 x 109 cells) yields at least 50,000-100,000 cells for each of the P1-hi, P1-low, and P2 populations with a viability of approximately 70%.
The key difference between the spleen- and BM-derived populations identified by this protocol is the much lower frequency of the P6 population and its subpopulations, P4/P5/P3, in spleen-derived cells.
As an example of flow cytometric analysis, the effect of Epo administration in mice was examined. Epo binds to and activates the Epo receptor (EpoR), which is essential for both basal and accelerated erythropoiesis under stress conditions like hypoxia. Hypoxia stimulates an increase in blood Epo levels28,18, in turn promoting the expansion of CFU-e1,29 and erythroid precursors.
Epo (0.25 U/g body weight) or an equal volume of vehicle (saline) were injected subcutaneously into mice once every 24 h for 3 days. The BM and spleen were harvested at 72 h. Epo stimulation led to the expansion of the early and late CFU-e populations (P1-low and P1-hi) in both the BM and spleen (Figure 4 and Figure 5).
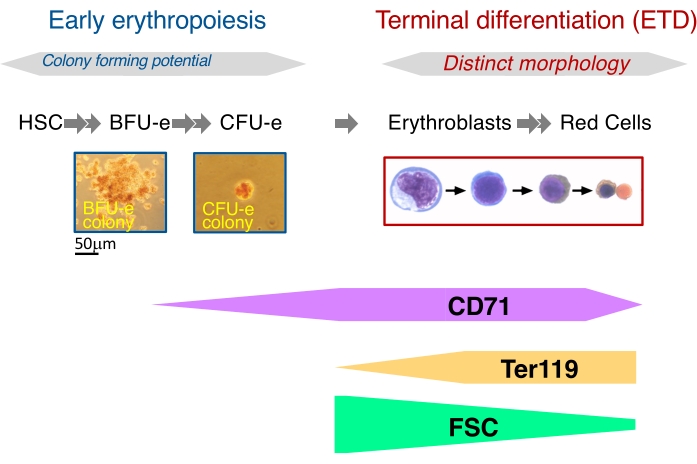
Figure 1: Two principal phases of definitive erythropoiesis. Erythropoiesis may be divided into an early phase, where progenitors are defined based on their colony-forming potential, and a later phase known as erythroid terminal differentiation (ETD), where the erythroblast stage is defined based on morphology. Flow cytometric techniques have permitted the prospective isolation of developmentally specific erythroblast stages using FSC, Ter119, and CD71. By contrast, there has not been an equivalent method for isolating BFU-e and CFU-e progenitors directly from tissue with high purity. Note that only a fraction of the BFU-e colony is shown. The scale bar applies to both the CFU-e and BFU-e colonies. Please click here to view a larger version of this figure.
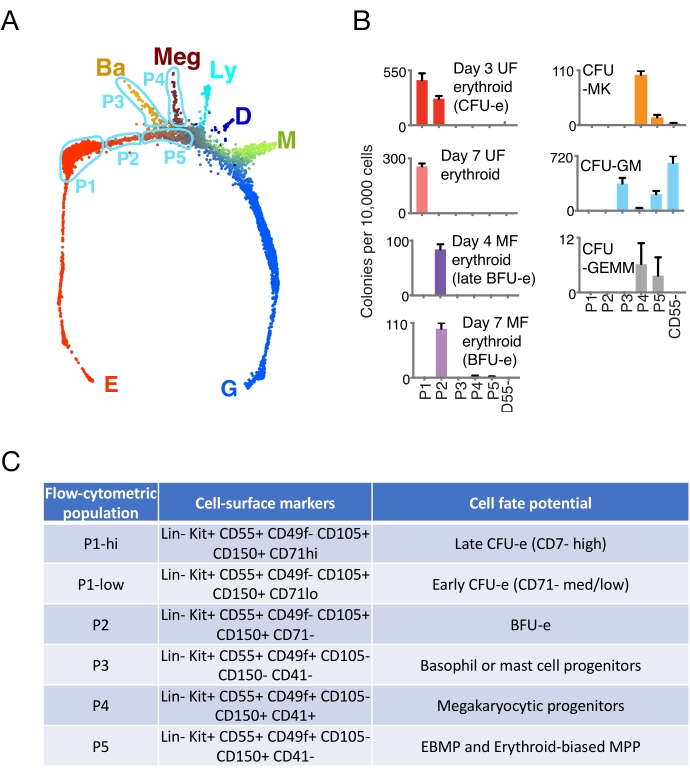
Figure 2: A new flow cytometric strategy for isolating CFU-e and BFU-e directly from tissue. This manuscript describes a protocol for identifying five new flow cytometric populations, P1 to P5, based on an analysis of the scRNAseq data. (A) Force-directed graph projection ("Spring plot") of fresh Balb/C adult bone marrow single-cell transcriptomes. The colors indicate the predicted cell fate potential based on transcriptomic information and the population-balance analysis (PBA) algorithm1. The regions of the plot that correspond to each of the flow cytometric populations P1 to P5 are indicated. The regions delineated are hand-drawn approximations of the original data, which may be found in Tusi et al.1. The correspondence was confirmed by undertaking scRNAseq of each of the sorted P1 to P5 populations1. E = erythroid; Ba = basophilic or mast-cell progenitors; Meg = megakaryocytic progenitors; Ly = Lymphocytic progenitors; D = dendritic progenitors; M = monocytic progenitors; G = granulocytic progenitors. (B) Colony formation assays, replotted from Tusi et al.1. The P1 to P5 populations and CD55+ cells were sorted as described in this protocol, and each population was plated for the relevant colony formation assays. Erythroid colonies were scored on the indicated days. UF = unifocal; MF = multi-focal; CFU-MK = colony-forming unit megakaryocytic; CFU-GM = colony-forming unit granulocytic/monocytic; CFU-GEMM = colony-forming unit granulocytic, erythroid, monocytic, megakaryocytic. (C) Cell surface markers that define each of the five populations, as well as their cell fate potential. Cell fate potential is both the result of transcriptomic information and of the cell fate assays, both colony formation and single-cell fate assays1,27. Of note, the P1 population was split based on the expression of the CD71 marker into P1-hi and P1-low1. Based on data from Tusi et al.1. Please click here to view a larger version of this figure.
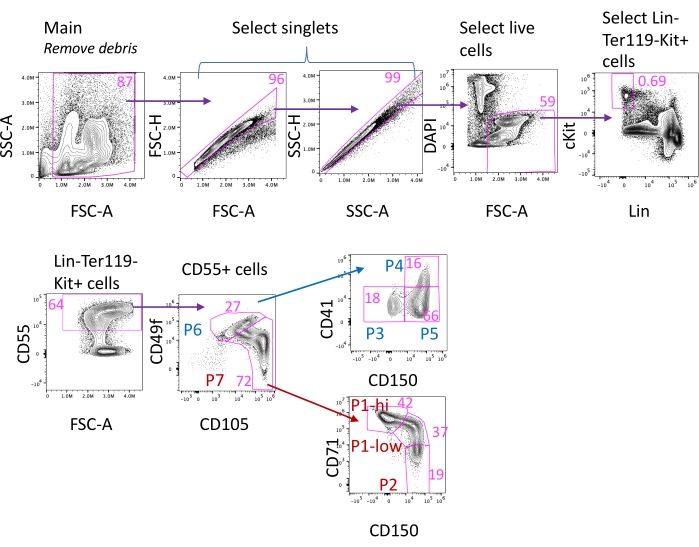
Figure 3: Flow cytometric gating strategy. The complete gating strategy for populations P1 to P5. Freshly harvested bone marrow (BM) or spleen (SP) from a Balb/C mouse subcutaneously injected with 100 µL of 0.9% sterile saline are first gated for the main population, discarding debris; this is followed by selecting singlets, discarding dead cells, and gating on cells that are Ter119-negative, lineage-negative, and express Kit. Further subdivision of Ter119–Lin–Kit+CD55+ into P6 and P7 is followed by the final subdivision into populations P1 to P5 (also see Figure 2). The Ter119 channel allows the analysis of erythroblasts on the same sample (using the Ter119/CD71 approach12). However, a separate Ter119 channel is not essential for this protocol, and the Ter119 antibody may be omitted from the master mix and included as part of the Lin-FITC cocktail instead. The exclusion step for Ter119 positive cells is not shown in this gating strategy. Numbers are percentages of each gate within the shown histogram. Please click here to view a larger version of this figure.
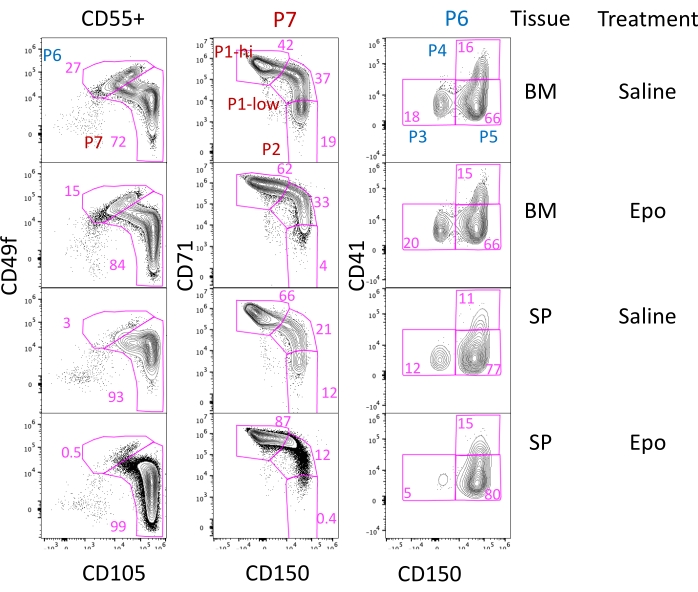
Figure 4: Analysis of bone marrow and spleen early erythroid progenitors following Epo stimulation. Representative flow cytometric plots of freshly harvested bone marrow (BM) or spleen (SP). Balb/C mice were injected subcutaneously with either 0.25 U/g body weight of Epo or with an equivalent volume of saline. Please click here to view a larger version of this figure.
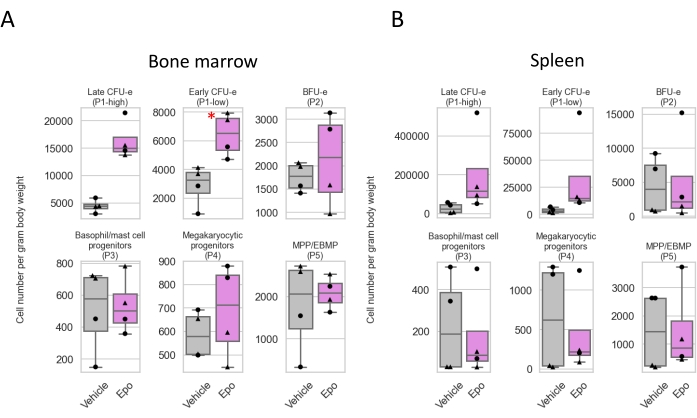
Figure 5: The response of BM and spleen progenitors to Epo in vivo. Summary statistics showing changes in the absolute cell number in populations P1 to P5 following Epo injection in freshly harvested (A) bone marrow and (B) spleen cells; *p = 0.02. Experiment as described in Figure 4. n = 4 mice in each group. To calculate the absolute cell number, the frequency of cells in each population in either the BM or spleen (from Figure 4) was multiplied by the total number of BM or spleen cells harvested from each mouse and divided by the mouse weight in grams. Of note, only early CFU-e in the BM reached statistical significance (p = 0.02), likely a combination of a relatively low Epo dose and a small number of mice in each group. Please click here to view a larger version of this figure.
| Detector Array | PMT | Dichroic mirror | Bandpass filter | Fluorophore detected |
| Blue Laser (488 nm) | A | 685LP | 695/40 | |
| B | 505LP | 530/30 | Lin-FITC (+Ter119-FITC) | |
| C | 488/10 | |||
| Violet Laser (405 nm) | A | 760LP | 800/80 | |
| B | 630LP | 660/20 | CD150-BV650 | |
| C | 595LP | 605/20 | CD41-BV605 | |
| D | 475LP | 525/20 | ||
| E | 450/50 | CD49f-BV421 | ||
| Red Laser (640 nm) | A | 755LP | 785/50 | cKit-APC-Cy7 |
| B | 700 LP | 720/30 | ||
| C | 675/14 | CD55-AF 647 | ||
| UV Laser (355 nm) | A | 505LP | 525/20 | |
| B | 420LP | 450/50 | DAPI | |
| C | 379/28 | Ter119-BUV 395 | ||
| YG Laser (561 nm) | A | 755LP | 780/60 | CD71-PE-Cy7 |
| B | 685LP | 710/50 | ||
| C | 635LP | 660/20 | ||
| D | 600LP | 610/20 | ||
| E | 570LP | 580/20 | CD105-PE |
Table 1: Example of the channel layout in an LSRII cytometer. The panel of antibodies used in the described protocol and their arrangement in the LSRII flow cytometer to collect the data.
| Reagent or Antibody | Conjugate | Stock conc. (mg/mL) | Dilution factor | Final conc. in cell suspension (µg/mL) | Clone |
| CD71 | PE/Cy7 | 0.2 | 2000 | 0.1 | RI7217 |
| TER-119 | BUV395 | 0.2 | 100 | 2 | TER-119 |
| CD55 (DAF) | AF647 | 0.5 | 50 | 10 | RIKO-3 |
| CD105 | PE | 0.2 | 50 | 4 | MJ7/18 |
| CD150 (SLAM) | BV650 | 0.1 | 50 | 2 | TC15-12F12.2 |
| CD49f | BV421 | 0.025 | 50 | 0.5 | GoH3 |
| CD41 | BV605 | 0.2 | 100 | 2 | MWReg30 |
| CD117 (cKit) | APC/Cy7 | 0.2 | 200 | 1 | 2B8 |
| Lin cocktail | FITC | 0.08 (for each antibody in the cocktail) | 83 | 1 (for each antibody in the cocktail) | |
| Rabbit IgG | 10 | 50 | 200 |
Table 2: Antibody master-mix components. Composition of the antibody master mix. The Ter119 channel allows the analysis of erythroblasts on the same sample (using the Ter119/CD71 approach12). However, a separate Ter119 channel is not essential for this protocol, and the Ter119 antibody may be omitted from the master mix and included as part of the Lin-FITC cocktail instead.
| Reagent or Antibody | Conjugate | Stock conc. (mg/mL) | Clone |
| Ly-6G and Ly-6C | FITC | 0.5 | RB6-8C5 |
| CD11b | FITC | 0.5 | M1/70 |
| CD19 | FITC | 0.5 | 1D3 |
| CD4 | FITC | 0.5 | RM4-5 |
| CD8a | FITC | 0.5 | 53-6.7 |
| F4/80 | FITC | 0.5 | BM8 |
Table 3: Lin cocktail components. Composition of the Lin cocktail used as part of the antibody master mix (see details of the master mix in Table 2).
| Reagent or Antibody | Conjugate | Stock conc. (mg/mL) | Dilution factor | Final conc. in cell suspension (µg/mL) | Clone |
| Ly-6G and Ly-6C | Biotin | 0.5 | 200 | 2.5 | RB6-8C5 |
| CD11b | Biotin | 0.5 | 200 | 2.5 | M1/70 |
| CD19 | Biotin | 0.5 | 200 | 2.5 | 1D3 |
| CD4 | Biotin | 0.5 | 200 | 2.5 | RM4-5 |
| CD8a | Biotin | 0.5 | 200 | 2.5 | 53-6.7 |
| F4/80 | Biotin | 0.5 | 200 | 2.5 | BM8 |
| TER-119 | Biotin | 0.5 | 200 | 2.5 | TER-119 |
| Normal rat serum | 50 |
Table 4: Antibodies for magnetic-bead enrichment. Antibodies used to enrich the Lin– fraction of either the BM or spleen prior to cell sorting.
