The absolute number of circRNAs in each sample can be derived from the exported dd-PCR data. Real-time quantitative PCR analysis suggested differential expression of circBnc2 in the differentiated C2C12 myotubes (data not shown). Here, we wanted to check the absolute copy number of circBnc2 in proliferating C2C12 myoblasts and myotubes. Since the expression of circBnc2 is compared in two conditions, it is really important to process all samples for RNA isolation, cDNA synthesis, and PCR simultaneously using the same reagents and procedure. To perform the absolute quantification of circRNAs using the dd-PCR reaction, an equal amount of initial total RNA should be used for cDNA synthesis to calculate the exact number of target RNA molecules per nanogram of total RNA. For example, 1 µg of total RNA was used for cDNA synthesis and diluted to 500 µL using nuclease-free water to get a final concentration of 2 ng/µL before performing the dd-PCR assay (Figure 1).
As shown in Figure 2A, cDNA from 10 ng of RNA from proliferating C2C12 myoblasts cells (MB) and a 4-day differentiated myotube (MT) was used to check the difference in expression of circBnc2 in these two conditions. The samples were processed to generate the droplet and perform PCR, followed by counting the positive and negative droplets using the droplet analysis software as per the manufacturer's instructions (Figure 2B). Since primers can amplify non-specific products and form primer dimers, NTC should be used for all primer sets. Ideally, NTC should have no positive droplet counts.
As shown in Figure 3A, all samples except MT1 showed more than 12,000 droplet counts. Since the total droplet count was low in MT1, this sample was not considered for final data analysis. The low total droplet count could be due to an error in droplet generation, rupture of the droplets during transfer to the PCR plate, or pipetting issues. Interestingly, there was a clear difference in the expression pattern of circBnc2 in the 4-day differentiated myotube condition compared to the myoblast cells. The analysis of circBnc2 abundance in terms of copy number indicated that the three replicates of C2C12 myoblasts had 76.8, 67, and 46 copies/ng of RNA, while the two myotube samples had 558 and 610 copies/ng of RNA (Figure 3B). Since one myotube sample did not work well, it is better to have a greater number of biological replicates to study the statistical differences in their expression patterns in the two conditions.
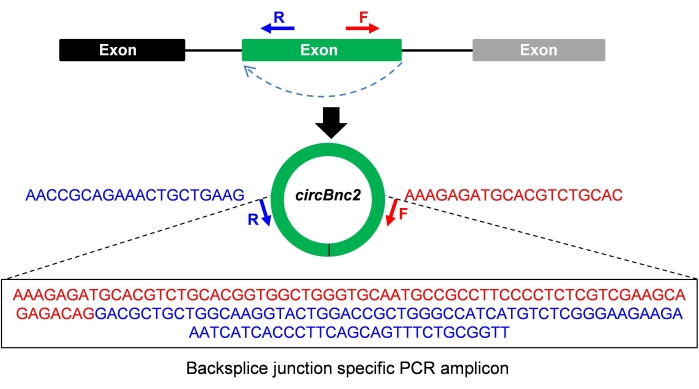
Figure 1: Schematic representation of the divergent primer design for the PCR amplification of circBnc2. Please click here to view a larger version of this figure.
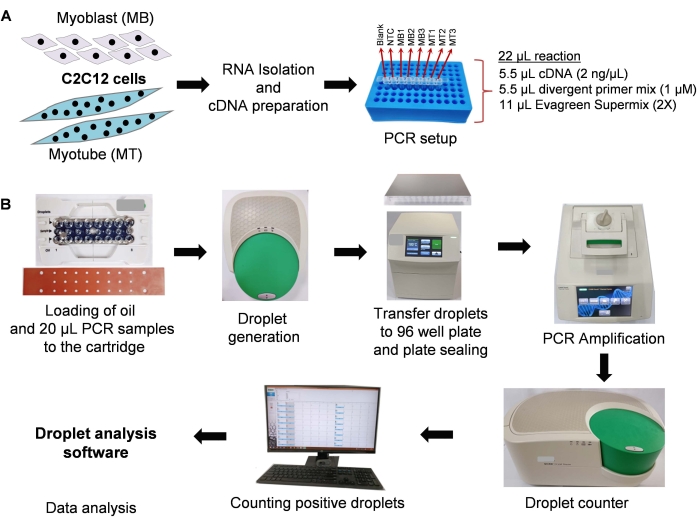
Figure 2: The dd-PCR workflow for circRNA quantification. The complete dd-PCR workflow includes many steps: (A) RNA isolation and cDNA preparation from C2C12 myoblast and 4-day differentiated myotube cells, PCR mixture preparation, (B) droplet generation, PCR amplification, droplet counting, and data analysis. Please click here to view a larger version of this figure.
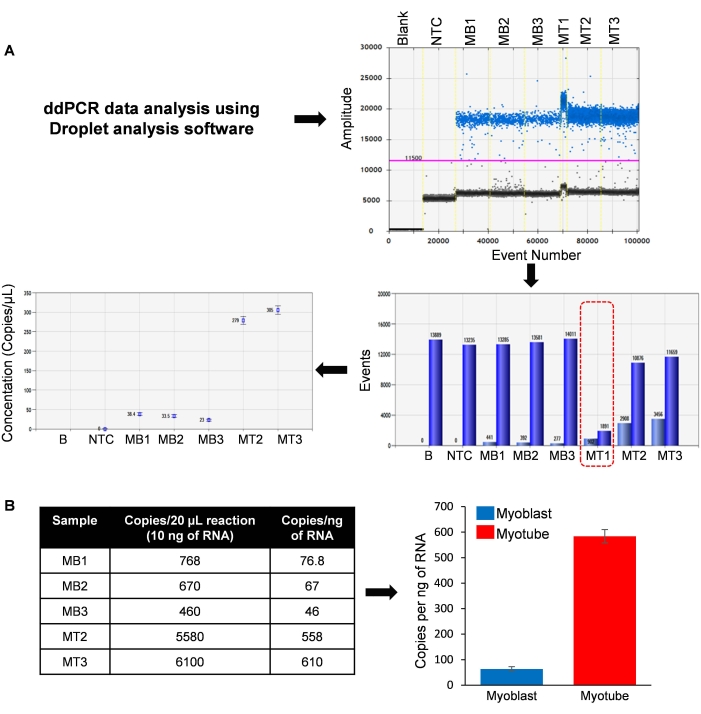
Figure 3: Data analysis using QuantaSoft software. (A) Data analysis includes the identification of positive and negative droplets using the quantitation software. (B) Calculating the number of circRNAs/ng of RNA in C2C12 myoblasts (MB) and myotubes (MT). The data in the panel B bar graph represent the mean ± STDEV of two to three biological replicates. Please click here to view a larger version of this figure.
| Primer Name | Sequence |
| circBnc2 Forward primer | AAAGAGATGCACGTCTGCAC |
| circBnc2 Reverse primer | AACCGCAGAAACTGCTGAAG |
Table 1: Divergent primer sequences used for the amplification of circBnc2.
| Step | Temperature (˚C) | Time | No of cycles | Ramp rate |
| Enzyme activation | 95 | 10 min | 1 | ~ 2 ˚C/s |
| Denaturation | 94 | 30 s | 40 | ~ 2 ˚C/s |
| Annealing/Extension | 60 | 1 min | 40 | ~ 2 ˚C/s |
| Signal stabilisation | 4 | 5 min | 1 | ~ 2 ˚C/s |
| Signal stabilisation | 90 | 5 min | 1 | ~ 2 ˚C/s |
Table 2: Conditions for PCR amplification of circBnc2 on a thermal cycler.
