Figure 2 illustrates the typical results of a SE-FRET experiment, in which the cell nuclei were simultaneously recorded in three channels (i.e., donor GFP, acceptor mRFP, and SE-FRET). These data were used to generate images of SE-FRET efficiency coded in a pseudo-color scale. On this scale, the transition from blue to red corresponds to an increase in FRET efficiency, a measure of protein-protein proximity from 0% to 100%. In this representative experiment, the SE-FRET signal was recorded in the cell nucleus, and its intensity following the coexpression of LSH10 and OTLD1 was comparable to that observed after the expression of the mRFP-GFP (i.e., positive control). No SE-FRET was observed in negative controls (i.e., coexpression of OTLD1-mRFP and LSH4-GFP or free mRFP and LSH10-GFP).
The LSH10-OTLD1 interactions were quantified using AB-FRET. To this end, the donor GFP fluorescence was recorded in the cell nucleus before and after the photobleaching of the acceptor mRFP as photobleaching time series of donor and acceptor fluorescence measurements (Supplementary Figure 4). The images of the recorded cell nuclei were presented in pseudo-color to quantify the change in GFP fluorescence. Figure 3 shows that the LSH10-GFP/OTLD1-mRFP coexpression resulted in an increased GFP donor fluorescence after the mRFP acceptor was photobleached and lost its ability to fluoresce. A similar increase in the donor fluorescence was observed in the mRFP-GFP positive control but not in the negative controls of LSH4-GFP/OTLD1-mRFP or LSH10-GFP/mRFP coexpression, whereas the acceptor fluorescence was inactivated in all photobleaching experiments. Figure 4 shows the quantitative analysis of the AB-FRET data, demonstrating the statistically significant increase in the donor fluorescence (%AB-FRET) of approximately 13% after coexpressing LSH10 and OTLD1. The positive mRFP-GFP control produced %AB-FRET of approximately 30%, whereas the negative controls produced no %AB-FRET. Both SE-FRET and AB-FRET images showed the FRET signal in the cell nucleus, consistent with the subcellular localization expected for the transcription factor-histone-modifying enzyme complexes as well as for the nucleocytoplasmic nature of the GFP/mRFP proteins34 (Figure 2 and Figure 3).
In summary, the representative data show that this FRET protocol can be used to demonstrate and quantify interactions between histone-modifying enzymes and transcription factors and determine their subcellular localization in living plant cells.
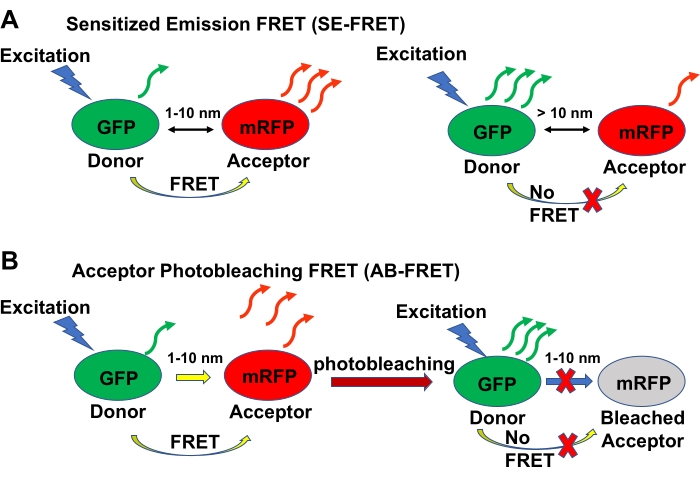
Figure 1: Schematic summary of the SE-FRET and AB-FRET techniques. (A) The basic principle of SE-FRET. One of the tested proteins is tagged with GFP, which acts as a donor fluorochrome, and the other with mRFP, which acts as an acceptor fluorochrome. The donor molecule is excited, and the acceptor emission is recorded. If the tested proteins interact with each other such that they are positioned within 10 nm of each other, the energy from the excited donor is transferred non-radiatively to the acceptor, which then becomes excited and emits fluorescence in the FRET emission channel. If no interaction occurs, no energy is transferred from the donor to the acceptor, and no FRET emission by the acceptor is detected. (B) The basic principle of AB-FRET. The tested proteins are tagged as described in (A) for SE-FRET. The donor molecule is excited, and if the interaction between the tested proteins occurs, the donor excites the acceptor in a non-radiative fashion, resulting in FRET. Then, the acceptor is permanently inactivated by photobleaching, thereby losing its ability to accept non-radiative energy from the donor and emit the FRET fluorescence in the FRET emission channel; the fluorescence emitted by the donor, on the other hand, is increased because the donor loses less energy by the non-radiative transfer. Please click here to view a larger version of this figure.
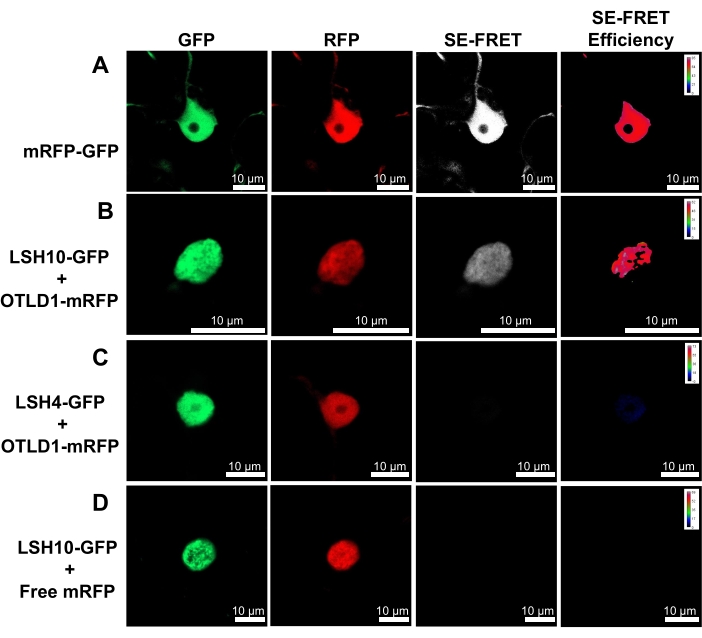
Figure 2: Specific interaction of LSH10 with OTLD1 in N. benthamiana leaves detected by SE-FRET. Images from three detection channels (donor, acceptor, and SE-FRET) are shown for the indicated protein combinations. The SE-FRET efficiency images were calculated by the subtraction of spectral bleed-through (SBT) and are shown in pseudo-color, with the colors red and blue signifying the highest and the lowest signal, respectively. (A) High SE-FRET efficiency signal produced by the mRFP-GFP positive control. (B) Positive SE-FRET efficiency signal produced by the interacting LSH10-GFP and OTLD1-mRFP proteins. (C) Coexpression of the negative control protein LSH4-GFP and OTLD1-mRFP produced no SE-FRET efficiency signal. (D) Coexpression of the negative control-free mRFP protein and LSH10-GFP produced no SE-FRET efficiency signal. Scale bars = 10 µm. Please click here to view a larger version of this figure.
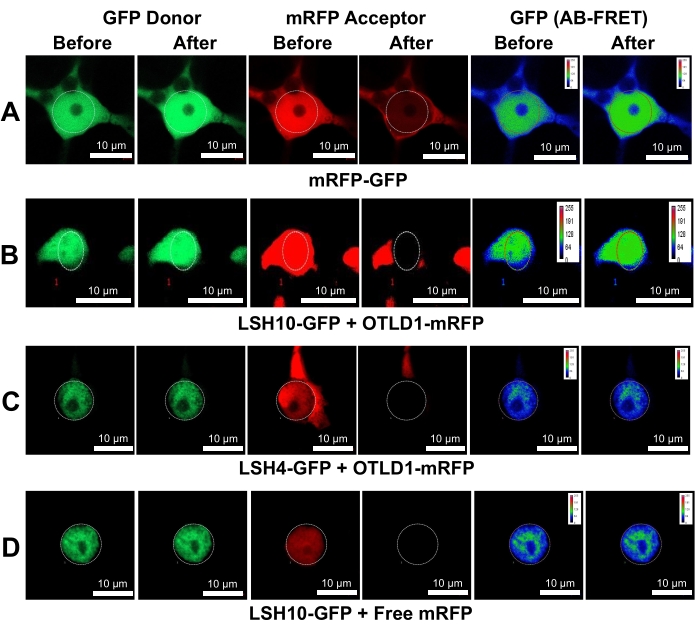
Figure 3: Specific interaction of LSH10 with OTLD1 in N. benthamiana leaves detected by AB-FRET. Images from two detection channels (donor and acceptor) before and after photobleaching are shown for the indicated protein combinations. The circle indicates the photobleached region. AB-FRET, visualized as an increase in GFP fluorescence after mRFP photobleaching, is displayed using pseudo-color with the colors red and blue, signifying the highest and lowest signal, respectively. (A) An increase in the GFP donor fluorescence produced by the mRFP-GFP positive control. (B) An increase in the GFP donor fluorescence produced by the interacting LSH10-GFP and OTLD1-mRFP proteins. (C) Coexpression of the negative control protein LSH4-GFP and OTLD1-mRFP produced negligible changes in the GFP donor fluorescence. (D) Coexpression of the negative control free mRFP protein and LSH10-GFP produced negligible changes in the GFP donor fluorescence. Scale bars = 10 µm. Please click here to view a larger version of this figure.
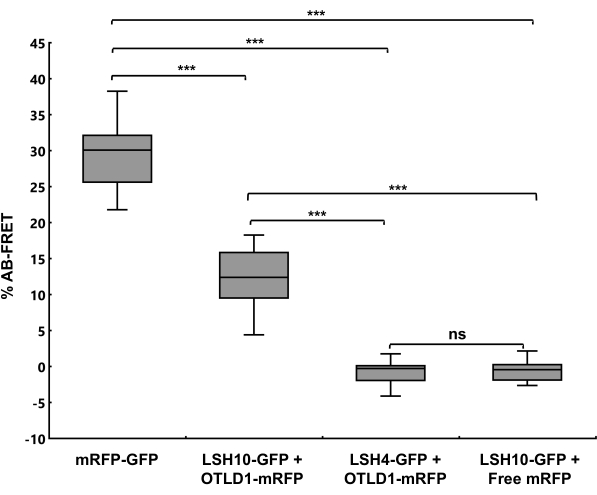
Figure 4: A Quantification of AB-FRET. The percentage increase in the GFP donor fluorescence after mRFP photobleaching (%AB-FRET) is shown for the indicated protein combinations. Error bars represent the mean for n = 13 cells for each measurement. The two-tailed t-test determined that differences between mean values are statistically significant for the p-values *p < 0.05, **p < 0.01, and ***p < 0.001; p≥ 0.05 are not statistically significant (ns). Please click here to view a larger version of this figure.
| Primer name | Sequence (5ʹ to 3ʹ) | Purpose | |||||
| OTLD1 Fw | ggggacaagtttgtacaaaaaagcaggctcaatgactcggattttggttcaaag | Amplify OTLD1 from cDNA | |||||
| OTLD1 Rv | ggggaccactttgtacaagaaagctgggtgttccgtggctttgcctttgcgtc | Amplify OTLD1 from cDNA | |||||
| LSH10 Fw | ggggacaagtttgtacaaaaaagcaggctcaatgtcctctccaagagaaagagg | Amplify LSH10 from cDNA | |||||
| LSH10 Rv | ggggaccactttgtacaagaaagctgggtgatgtcaacagagactaaagaaac | Amplify LSH10 from cDNA | |||||
| LSH4 Fw | ggggacaagtttgtacaaaaaagcaggctcaatggatcatatcatcggctttatg | Amplify LSH4 from cDNA | |||||
| LSH4 Rv | ggggaccactttgtacaagaaagctgggtgattagggctacttgaaatcgcc | Amplify LSH4 from cDNA | |||||
| mRFP Fw | ggggacaagtttgtacaaaaaagcaggctcaatggcctcctccgaggacgt | Amplify mRFP from pPZP-RCS2A-DEST-mRFP-N1 | |||||
| mRFP Rv | ggggaccactttgtacaagaaagctgggtgttggagatctgcggccgcgg | Amplify mRFP from pPZP-RCS2A-DEST-mRFP-N1 | |||||
| AttL1 | tcgcgttaacgctagcatggatctc | Confirm sequences in pDONR207 by PCR and DNA sequencing | |||||
| AttL2 | gtaacatcagagattttgagacac | Confirm sequences in pDONR207 by PCR and DNA sequencing | |||||
| AttB1 Fw | ggggacaagtttgtac aaaaaagcaggct | Confirm sequences in destination vectors by PCR and DNA sequencing | |||||
| AttB2 Rv | ggggaccactttgta caagaaagctgggt | Confirm sequences in destination vectors by PCR and DNA sequencing | |||||
| 35S Promoter Fw | ctatccttcgcaagacccttc | Confirm sequences in destination vectors by PCR | |||||
Table 1: Primers for cloning and confirming the cloned sequences in pDONOR207 and destination vectors. Fw, forward primers; Rv, reverse primers.
Supplementary Figure 1: Setting parameters for confocal channels. (A) Screenshot for the excitation and emission parameter setup for the donor channel (GFP). (B) Screenshot for the excitation and emission parameter setup for the acceptor channel (mRFP). (C) Screenshot for the excitation and emission parameter setup for the FRET channel. Please click here to download this File.
Supplementary Figure 2: Adjusting parameters for the acquisition of SE-FRET images of the sample of interest. (A) Screenshot for the scan area parameter setup (i.e., image size, scan speed, direction, and averaging). (B) Screenshot for the GFP channel parameter setup (i.e., laser, pinhole, master gain, and digital gain). (C) Screenshot for the mRFP channel parameter setup (i.e., laser, pinhole, master gain, and digital gain). (D) Screenshot for the FRET channel parameter setup (i.e., laser, pinhole, master gain, and digital gain). Please click here to download this File.
Supplementary Figure 3: Setting parameters for the acceptor photobleaching. (A) Screenshot for the scan area parameter setup (i.e., image size, scan speed, direction, and averaging). (B) Screenshot for the time series and time bleaching parameter setup. Please click here to download this File.
Supplementary Figure 4: Time series of the donor and acceptor fluorescence measurements during AP-FRET. The kinetics of the acceptor (mRFP) and donor (GFP) fluorescence was determined for the indicated samples before, during, and after the photobleaching period. (A) Positive mRFP-GFP control. (B) LSH10-GFP + OTLD1-mRFP. (C) Negative LSH4-GFP + OTLD1-mRFP control. (D) Negative LSH10-GFP + Free mRFP control. Yellow lines indicate the photobleaching time period. White curves plot the measurements of the fluorescence kinetics. In each panel, the upper and the lower images show the kinetics of the acceptor (mRFP) and donor (GFP) fluorescence, respectively. Note that, naturally, the GFP fluorescence often decreases over time because the laser gradually photobleaches the GFP itself. Please click here to download this File.
