1. Establishing a low-noise system
NOTE: The relevant experiments require a potentiostat capable of achieving the highly time-resolved measurement of low currents. To achieve this, employ a research-grade commercial potentiostat capable of a 1 µs time resolution that can quantify currents at the femtoampere level. To further reduce electronic interference from the environment, conduct experiments within two nesting Faraday cages. Ensure that the setup is capable of a root-mean-square deviation of less than 100 fA for a chronoamperometry experiment sampled at 10 Hz in 0.1 M potassium chloride.
- Obtain and set up the equipment, including the potentiostat and Faraday cages.
NOTE: Faraday cages may be obtained commercially or custom-fabricated using conductive metals (e.g., copper or aluminum). Custom-fabricated aluminum Faraday cages were used for the study described here (see Table of Materials).
2. Experimental preparation
- Use commercially available 2 µm diameter carboxylate-modified polystyrene beads (see Table of Materials).
NOTE: While this system can be generalized to other electro-inactive species23,33, it is critical to remember that the impacts rely on electrophoretic migration in addition to Brownian motion. Thus, apply a potential attractive to the species of interest, and maintain low salt concentrations17. - Prepare the following solutions, which can be stored at room temperature for at least 1 month.
- Prepare a 50 mM carbonate solution, and titrate to pH 12.0 using 1 M NaOH. Monitor the pH weekly.
- Prepare a 1 M sodium perchlorate solution.
- Prepare the following solutions fresh each day, discarding them at the end of the day.
- Prepare a 10 mM TEMPO in 50 mM carbonate solution, pH 12.0.
- Prepare a 500 mM maltose stock in 50 mM carbonate solution, pH 12.0.
- Select the working electrode. For consistent results, select an ultramicroelectrode (see Table of Materials) such that the radius of the species to be characterized is no less than 10%-15% of the electrode radius17,21,23,33,34.
NOTE: This ratio can be minimized after determining the magnitude of the impact-associated current steps for the optimal detection of a particular species of interest. The electrode material selected must catalyze the background redox reaction. - Prepare two electrochemical cells of 5 mL each.
- Prepare a control cell containing 1 mM TEMPO and 5 mM sodium perchlorate (see Table of Materials) in carbonate buffer at pH 12.0.
- Prepare a test cell containing a solution of 1 mM TEMPO, 5 mM sodium perchlorate, and 120 mM maltose in carbonate buffer at pH 12.0.
NOTE: The ratio of the redox mediator to the substrate (here, TEMPO to maltose) can be varied to explore the effects of the reaction rate20. This value provides insight into the homogeneous chemical reaction. - After preparing these cells, set them aside for later electrochemical measurements.
3. Electrode polishing
- Prior to each experimental run, polish the electrode sequentially for 2 min each with a 1 µm, 0.3 µm, and 0.5 µm alumina slurry (see Table of Materials) on polishing pads.
- Move the electrode in a "figure 8" pattern to ensure an even polish35,36. Liberally rinse with deionized water, and dry with a laboratory wipe.
NOTE: Do not sonicate the ultramicroelectrodes, as this may damage them.
4. Electrochemical measurements
NOTE: See Figure 2 for the results.
- Utilize a three-electrode setup for the electrochemical measurements. For the experiments described here, employ an 11 µm carbon-fiber ultramicroelectrode, a platinum wire counter electrode, and a saturated calomel reference electrode (SCE) (see Table of Materials).
NOTE: Electrochemical potential windows for the experiments are noted below; for reference, TEMPO has a formal potential of 0.49 V versus SCE, and maltose is not electroactive in the potential window used in these experiments. Any leaking from the reference electrode may affect the total salt concentration37, thus reducing the electrophoretic driving of the particles to the ultramicroelectrode and reducing the counting efficiency17. If the experiments yield a low impact frequency, consider switching to a leakless reference electrode38,39. - Set the control cell in the Faraday cages, and connect the electrodes to the appropriate cables. Collect the first set of electrochemical measurements for each cell. This will consist of a cyclic voltammetry experiment and a chronoamperometry experiment, as detailed below.
- Collect the cyclic voltammetry data using a potential window from 0.2 V to 0.8 V versus SCE at scan rates of 10 mV·s−1, 20 mV·s−1, 30 mV·s−1, 40 mV·s−1, and 50 mV·s−1.
- Collect the chronoamperometry data by applying 0.8 V versus SCE for 10 min, and record at a 10 Hz sampling rate.
NOTE: A 10 min sampling period is recommended for chronoamperometry experiments to obtain 10-15 individual impacts at later steps. No impacts are expected at this juncture.
- Next, spike a dilute solution of polystyrene beads to a final concentration of 0.66 pM17 into each of the electrochemical cells. After the addition of the polystyrene beads, collect the second set of electrochemical measurements for each cell.
NOTE: The impact frequency will be a function of this concentration and requires optimization to collect sufficient data for statistical analysis without saturating the chronoamperogram with overlapping impacts40,41,42,43.- Collect chronoamperometry data by applying 0.8 V versus SCE for 10 min, and record at a 10 Hz sampling rate.
- Repeat the chronoamperometric measurements until sufficient data points have been collected for statistical analysis. To detect differences between the multiple sizing methods with a confidence level of 95% and a power of 80%, select a sample size of approximately 200 individual impact events.
5. Scanning electron microscopy (SEM)
NOTE: Use scanning electron microscopy as a "gold standard" technique to confirm the nanoparticle sizes and sample heterogeneity19,44.
- Sample preparation
- To prepare a sample for imaging, dilute a carboxyl latex bead suspension (see Table of Materials) 1:20 in water. Drop-cast 10 µL onto a glass slide, dry under a nitrogen stream, and sputter-coat the sample with a conductive layer of gold-palladium under argon.
- Imaging
- Using an accelerating beam voltage of 5 kV and a current of 0.4 nA, collect images as appropriate for statistical analysis. Use ImageJ45,46 or an equivalent image analysis software to determine the particle sizes.
6. Electrochemical data analysis
- Record the electrochemical data using the potentiostat's software, and analyze these results using a written script20 that can extract the current magnitudes from the detected changes in the steady-state current (current steps) resulting from nanoimpact events20.
NOTE: This script is included as part of the supplemental information in our previously published report20. - Convert the amplitudes of the current steps to bead radii using the following equation:
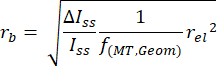
NOTE: Here, rb is the bead radius, rel is the electrode radius, ΔIss/Iss is the ratio between the change in the current produced by a particle's adsorption and the initial steady-state current observed prior to the adsorption of that particle, and f(MT,Geom) = 0.067 is an empirical scaling factor that depends on both geometric and mass transport considerations20,25. - Plot the frequency of detecting a given bead radius versus the radius (Figure 3) to quantify the distribution metrics.
7. Modeling
NOTE: If desired, the mechanism by which electrocatalytic interruption works can be validated by confirming the shift from the diffusion-limited current generation to the reaction rate-limited current generation. To describe and visualize, use two different numerical simulation programs: a voltammogram fitting software, such as DigiSim, to determine the homogeneous rate constant, and a multiphysics modeling platform, such as COMSOL Multiphysics, to visualize the local changes to the diffusion profile at the ultramicroelectrode surface (see Table of Materials).
- Voltammogram fitting
NOTE: Use the voltammogram fitting software to determine the homogeneous rate constant (Figure 4).- Collect cyclic voltammograms in a solution containing 1 mM TEMPO (only) and 1 mM TEMPO plus 120 mM maltose. For each condition, collect data at various scan rates, and use this data for numerical fittings of these experimental datasets.
- Fit the voltammograms collected from the TEMPO-only experiment using an E mechanism, which describes a reaction process driven solely by the electrode47,48. This will yield the electrode parameters.
- Using the electrode parameters obtained from step 7.1.1.1, fit the resulting voltammograms from the 1 mM TEMPO plus 120 maltose solution to an EC′ mechanism47,48, which describes an electrode process that is followed by a homogeneous chemical reaction that regenerates the redox mediator. This will yield the homogeneous rate constant.
- Collect cyclic voltammograms in a solution containing 1 mM TEMPO (only) and 1 mM TEMPO plus 120 mM maltose. For each condition, collect data at various scan rates, and use this data for numerical fittings of these experimental datasets.
- Multiphysics modeling
- Use a multiphysics modeling platform to visualize the changes in the diffusion profiles at the ultramicroelectrode surface for both the control and electrocatalytic interruption systems20,49,50,51,52 (Figure 5). Use the electrode parameters and homogeneous rate constant obtained from the voltammogram fitting as initial conditions. A broad overview of the workflow, which can be adapted to different software, is provided below.
- Input the global parameters. These consist of fixed values such as (but not limited to) the concentration values, diffusion coefficients, electrode radius, and temperature.
- Build the simulation space. This is a set of geometries that include the electrode, the insulating sheath, the surrounding solution space representing the region of interest, and an infinite element domain representing the bulk surroundings.
- Introduce physics packages to define the simulation.
- Associate the electrode space with an electroanalysis study. Define the initial values and electrode reaction of interest here.
- Associate the surrounding solution space with a chemistry study. Define the homogeneous chemical reaction that follows the electrode reaction here.
- Introduce a mesh over the entire simulation space. This defines how the geometry is divided to solve the model. To achieve high-quality results, use a finer mesh near the electrode.
- To observe changes as a result of the homogeneous rate constant, vary the value of this parameter using a parametric study to solve the model.
- To observe changes as a function of time, vary the value of this parameter using a time-dependent study to solve the model.
Electrocatalytic interruption mitigates edge effects by shifting the primary current generation mechanism from diffusion-limited (i.e., limited by the transport of a redox probe to the electrode) to kinetically limited (i.e., limited by a rapid, solution-phase reaction)20. This method is modular, meaning it allows a mix-and-match approach to choosing the electrode material, redox probe, and substrate, and this renders electrocatalytic interruption amenable to the detection of many nano- and bio-materials6,7,8,9,10,11,12,13,14,15,22. Implementing this technique on a 5.5 µm radius carbon-fiber electrode produced a 10-fold improvement in the precision associated with the electrochemical sizing of a model system (polystyrene beads) in a solution containing TEMPO as a redox probe and maltose as a substrate.
Following this protocol, the data sets required to validate this mechanism and its ability to restore analytical precision when sizing electro-inactive nanoparticles can be obtained. First, the cyclic voltammogram data collected in the absence of polystyrene beads showed a reversible redox event in control experiments involving TEMPO alone. From here, the addition of maltose resulted in an increase in the oxidative peak and a concurrent loss in the reductive peak as the oxidized TEMPO was regenerated by maltose. Second, the chronoamperograms collected under these conditions demonstrated that the steady-state currents at an oxidative potential were higher, consistent with the catalytic amplification observed in the cyclic voltammetry results. This step also suggests that the bulk chemical reaction is maintained by the electrode reaction; thus, any improvements over the control method will last over the measurement duration. However, this alone is insufficient for assessing any improvements to the measurement precision; to do so, chronoamperometry data must be collected in the presence of polystyrene beads.
To assess the sizing precision, chronoamperogram data were collected using 2 µm carboxylated polystyrene beads. Upon their addition, step-wise changes in the chronoamperogram current were observed as individual particles impacted and absorbed (Figure 2A control, Figure 2B electrocatalytic interruption). Each step-wise change in the steady-state current magnitude was converted to particle radii, and the data were visualized as histograms to compare the distribution from these electrochemical techniques to that of a gold standard technique, such as scanning electron microscopy (Figure 3). This comparison then allowed the characterization of precision metrics associated with each sizing approach.
Modeling was utilized to support these experimental observations. Specifically, fitting the cyclic voltammograms from before yielded parameters that characterized both the electrode reaction and the solution-phase chemical reaction (Figure 4). From the control solution, some sample parameters obtained were Efθ = 0.49 V, k0 = 0.02 cm·s-1, and ν = 10 mV·s-1 at T = 25° C. From the test solution, the kinetic parameters that limited current generation could be obtained; specifically, as Keq approaches infinity, kobs = 2,200 M-1·s-1. The numerical simulations could then use these values as the initial conditions for generating a concentration profile of the redox probe (Figure 5). In the absence of maltose, the resultant diffusion profile was radial, leading to heterogeneous material flux; specifically, more material diffused to the electrode at the edges. The introduction of maltose compressed the diffusion profile, producing, in turn, more homogeneous currents across the electrode surface.
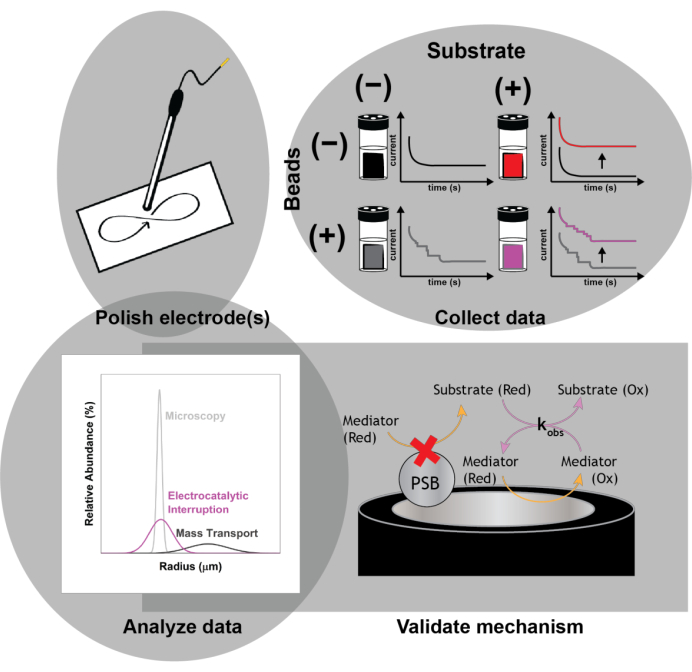
Figure 1: Schematic of the experimental protocol. Polish the electrodes prior to each experimental run. Collect a baseline set of electrochemical measurements (cyclic voltammetry and chronoamperometry) in the absence of beads with and without implementing electrocatalytic interruption to observe the current enhancement with the addition of the substrate. Spike in the beads, and collect a second set of electrochemical measurements for the size determination of the impacting nanoparticles. Validate the mechanism of action using numerical simulations. Please click here to view a larger version of this figure.
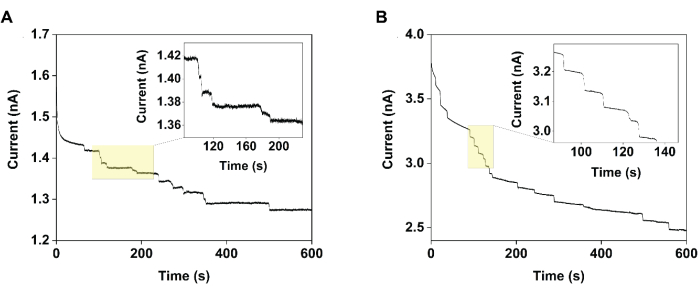
Figure 2: Chronoamperograms collected using an 11 µm diameter carbon-fiber ultramicroelectrode demonstrating the improvement in measurement precision achieved using electrocatalytic interruption. Specifically, when measuring the current versus time in a solution of 1 mM TEMPO in the (A) absence (control) and (B) presence of 120 mM maltose (electrocatalytic interruption), the steps observed in the latter case were more homogeneous. Reprinted with permission from Chung et al.20. Please click here to view a larger version of this figure.
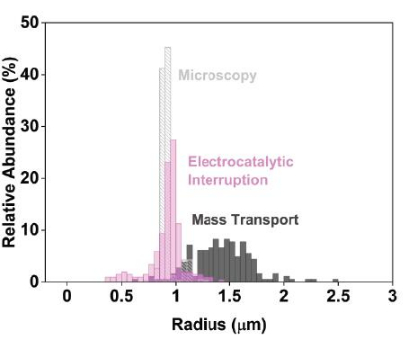
Figure 3: Precise electrochemical sizing data when using electrocatalytic interruption compared to the conventional, diffusion-limited electrochemical approach. To represent this data, prepare histograms comparing the size distributions determined using scanning electron microscopy (light grey) and electrochemistry (electrocatalytic interruption, pink; control, dark grey). Conventional nanoimpact studies, in which the current is limited by the mass transport of the mediator, produce artificially broad estimated size distributions (dark grey). In contrast, implementing electrocatalytic interruption leads to narrower, more precise size estimations (pink). Reprinted with permission from Chung et al.20. Please click here to view a larger version of this figure.
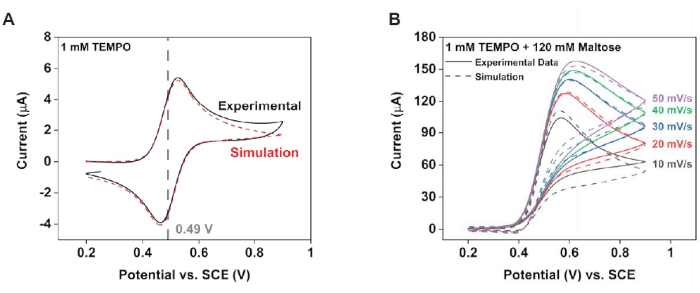
Figure 4: Modeling the electrode kinetics to characterize the new reaction scheme. Using cyclic voltammogram fitting software, extract the electrode reaction parameters from the experimental data. (A) Data with 1 mM TEMPO. (B) Data with 1 mM TEMPO plus 120 mM maltose. Reprinted with permission from Chung et al.20. Please click here to view a larger version of this figure.
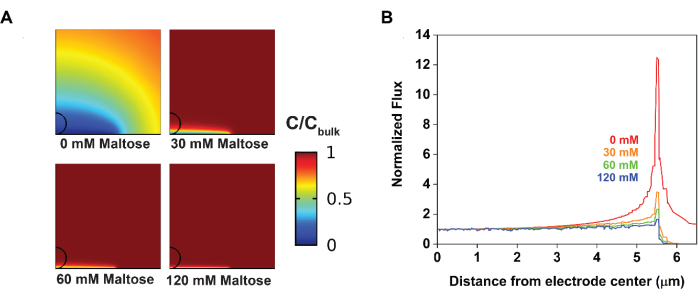
Figure 5: Changes in material flux at the electrode surface upon introducing electrocatalytic interruption visualized by numerical simulations. (A) The addition of maltose compresses the diffusion layer in a concentration-dependent manner. (B) The addition of maltose depresses the heterogeneous flux at the electrode edges. Reprinted with permission from Chung et al.20. Please click here to view a larger version of this figure.
