The protocol described here allows the efficient generation of cerebral organoids from human, chimpanzee, rhesus macaque, and common marmoset iPSC lines with minimal timing alterations required between species (Figure 1A). These organoids can be electroporated in the range of 20 dps to 50 dps, depending on the accessibility of the ventricle-like structures and the abundance of the cell population(s) of interest. However, prior to electroporation, it is important to determine whether the cerebral organoids are of sufficient quality to be electroporated.
A cerebral organoid ideal for electroporation should exhibit pronounced bright ventricle-like structures on the periphery, no signs of degeneration (e.g., detaching cells, enlarged apoptotic core), and a generally compact healthy morphology (e.g., no excessive outgrowth) (Figure 1B, "Go"). It is preferable to choose cerebral organoids with large, well-organized, ventricle-like structures to target a higher number of cells. If the peripheral zone of an organoid is dark and does not show any protruding structures, it is recommended to not use it for electroporation, as the precise microinjections might be compromised by the lack of visual cues (Figure 1B, "No-go"). To achieve an optimal cerebral organoid morphology, it is essential to ensure that critical steps such as the neuroectoderm induction and matrix embedding are well-timed. Problems concerning cerebral organoid morphology typically originate from failed neuroectoderm and/or neuroepithelial bud formation. This is normally caused by suboptimal timing of the neural induction and/or the basement membrane matrix embedding and can be solved by adjusting the timing of these steps (further troubleshooting tips for cerebral organoid formation can be found in Lancaster and Knoblich34).
After electroporation, a first assessment of its success and its efficiency can be conducted after 12 h, when the GFP expression of the transfected cells becomes detectable under a conventional inverted fluorescence microscope. Ideally, at this stage, multiple ventricle-like structures emit bright green fluorescence localized to one of their sides (Figure 2A). This indicates the high precision and efficiency of the procedure. Successfully electroporated cerebral organoids of the four different primate species (i.e., human, chimpanzee, rhesus macaque, and common marmoset) show similar GFP-positive patterns within the targeted ventricle-like structures (Figure 2A). Moreover, after the fixation and cryosectioning of electroporated primate cerebral organoids, successfully electroporated ventricle-like structures of all four species exhibit columns of GFP-positive cells within the radially organized and densely packed ventricular zone (VZ) (Figure 2B). The quantification of the DAPI-positive cells that were also GFP-positive in such regions of the chimpanzee and marmoset cerebral organoids 2 days post electroporation (17 ventricles from 12 organoids quantified) showed that, on average, roughly one-third of the cells (33%, SD ± 12%) were successfully electroporated.
Suboptimal electroporations are marked either by a small number of GFP-positive cells within the ventricle-like structure (Figure 3A) or by a few GFP-positive cells distant from any ventricle-like structure (Figure 3B). A low number of GFP-positive cells is caused by a poor plasmid uptake. This might be either due to a low plasmid concentration caused by an insufficient amount of microinjected electroporation mix or due to electric pulses that are not well-directed, which may be caused by suboptimal positioning of the cerebral organoids in the Petri dish electrode chamber. A low number of GFP-positive cells distant from any ventricle-like structure is caused by the electroporation of postmitotic cells within the cerebral organoid (e.g., neurons) due to an imprecise microinjection. These suboptimal electroporations need to be excluded from any further analyses.
The reliable identification of the cell types present in cerebral organoids is based, among other things, on the cell position within a ventricle-like structure, which requires a border definition between the VZ and SVZ/neuron-enriched zone. This border can be identified by the radial organization and high cell nuclei density characteristics of the VZ (see DAPI staining in Supplementary Figure S1). The confirmation of the VZ/SVZ border can be performed by immunofluorescence staining for neural progenitor markers such as PAX6 or SOX2, which are expressed by virtually all VZ cells (APs) and some SVZ cells (BPs). The presence of a neuron-enriched zone can be validated by immunofluorescence staining for neuronal markers such as class III β-tubulin (TUJ1) or NeuN (Supplementary Figure S1).
The duration of cerebral organoid culture post electroporation depends on the biological question and the cell populations of interest. In a recent study, it was demonstrated that different lengths of further culture after electroporation affect different cell populations in chimpanzee cerebral organoids, ranging from APs to upper-layer neurons24. Here, we show similar results for electroporated marmoset organoids. Specifically, 2 days after electroporation, GFP-positive cells are almost exclusively localized in the VZ and are also positive for PAX6—a marker for neural progenitor cells-indicating that these cells are APs or newborn BPs (Figure 4A). If the culture period after electroporation is extended to 10 days, then GFP-positive cells are localized in the basal regions (i.e., the SVZ and neuron-enriched zone) (Figure 4B,C). These cells can (in addition to the GFP signal) also be positive for PAX6 (Figure 4B), which is indicative of BPs, or NeuN (Figure 4C), which is indicative of neurons. Similar results can be obtained for human and rhesus macaque electroporated cerebral organoids. In summary, different progenitor types, as well as neurons, can be successfully targeted by this technique.
Almost all the previously shown data were derived from the immunostaining of histological sections generated from electroporated cerebral organoids. However, another elegant way to analyze these organoids is to perform whole-mount immunostaining followed by optical clearing35,36. This would allow 3D reconstruction of the electroporated cerebral organoids to obtain an impression of the 3D distribution of the GFP-positive cells. Figure 5 and Video 1 show a representative example of the GFP signal in an optical-cleared, electroporated cerebral organoid.
In summary, the electroporation protocol described here provides a precise and efficient way to introduce transient genetic modification(s) into different progenitor types and neurons of cerebral organoids derived from different primate iPSC lines.
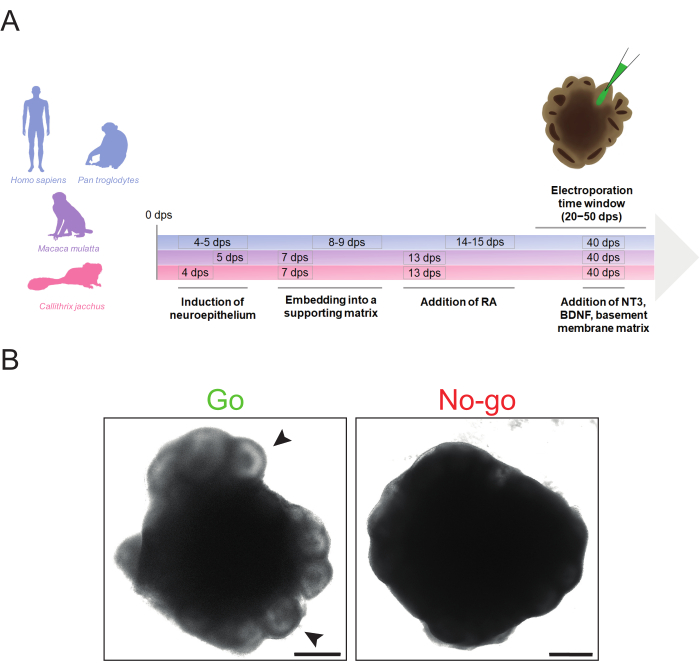
Figure 1: Schematic overview of primate cerebral organoid generation and morphological "go" and "no-go" criteria for electroporation. (A) Timeline of primate cerebral organoid generation and electroporation highlighting the different timings of the protocol steps for human and chimpanzee (blue), rhesus macaque (violet), and marmoset (magenta). Note that the chronology of the timeline is not to scale. (B) Brightfield images of a suitable (left image, Go) and an unsuitable (right image, No-go) 32 dps human cerebral organoid. The arrowheads indicate examples of suitable ventricle-like structures for microinjection. The images were acquired using a Zeiss Axio Observer.Z1 inverted fluorescence microscope with a 2.5x objective. Scale bars = 500 µm. Abbreviations: BDNF = brain-derived neurotrophic factor; dps = days post seeding; NT3 = neurotrophin 3; RA = retinoic acid. Please click here to view a larger version of this figure.
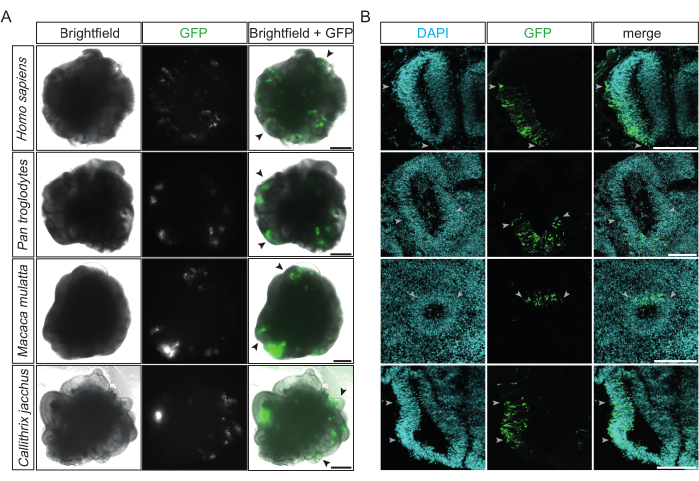
Figure 2: Examples of successfully electroporated primate cerebral organoids. (A) Brightfield (left column), fluorescence (middle column), and merge (right column) images of 22 dps human, 32 dps chimpanzee, 32 dps rhesus macaque, and 31 dps marmoset cerebral organoids (from top to bottom) 15-48 h after electroporation with the GFP-expressing plasmid. The black arrowheads indicate examples of individual electroporated ventricle-like structures. The images were acquired using a Zeiss Axio Observer.Z1 inverted fluorescence microscope with a 2.5x objective. Scale bars = 500 µm. (B) Immunofluorescence for GFP (green) combined with DAPI staining (cyan) of 32 dps human, 34 dps chimpanzee, 32 dps rhesus macaque, and 32 dps marmoset cerebral organoids (from top to bottom) 2-4 days after electroporation with the GFP-expressing plasmid. The light gray arrowheads indicate the borders of the electroporated regions within the ventricle-like structures. The images were acquired using a Zeiss LSM 800 confocal microscope with a 10x objective. Scale bars = 150 µm. Abbreviations: DAPI = 4',6-diamidino-2-phenylindole; dps = days post seeding; GFP = green fluorescent protein. Please click here to view a larger version of this figure.
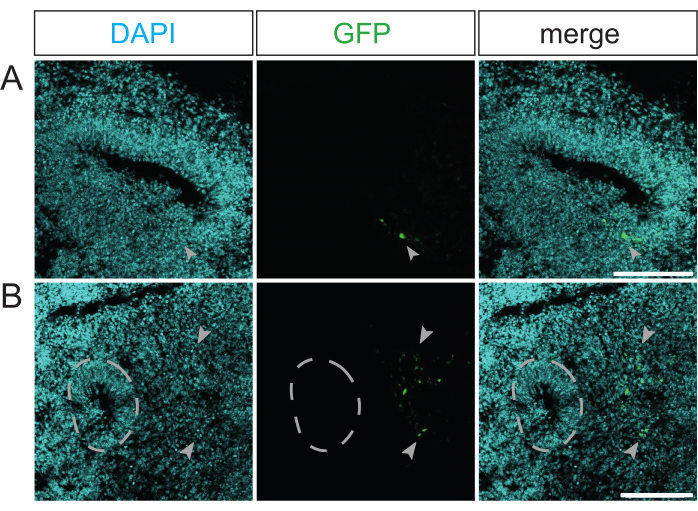
Figure 3: Examples of unsuccessfully electroporated primate cerebral organoids. (A,B) Immunofluorescence for GFP (green) combined with DAPI staining (cyan) of a (A) 34 dps rhesus macaque cerebral organoid 4 days after electroporation with the GFP-expressing plasmid and of (B) a 32 dps rhesus macaque cerebral organoid 2 days after electroporation with the GFP-expressing plasmid. The light gray arrowheads indicate electroporated cells. The light gray dashed outline indicates the border between the VZ and SVZ/neuron-enriched zone of a ventricle-like structure adjacent to the electroporated cells. The images were acquired using a Zeiss LSM 800 confocal microscope with a 10x objective. Scale bars = 150 µm. Abbreviations: DAPI = 4',6-diamidino-2-phenylindole; dps = days post seeding; GFP = green fluorescent protein; SVZ = subventricular zone; VZ = ventricular zone. Please click here to view a larger version of this figure.
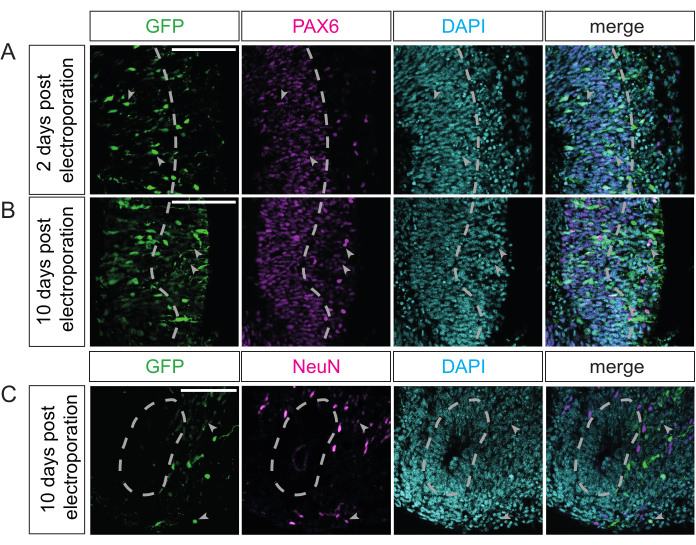
Figure 4: Visualization of the various cell populations present in primate cerebral organoids post electroporation. (A–C) Double immunofluorescence for GFP (green) and either PAX6 (A,B; magenta) or NeuN (C; magenta), in all cases combined with DAPI staining (cyan), of a (A) 32 dps marmoset cerebral organoid 2 days after electroporation with the GFP-expressing plasmid, and (B,C) of a 40 dps marmoset cerebral organoid 10 days after electroporation with the GFP-expressing plasmid. The light-gray arrowheads indicate (A,B) GFP+ and PAX6+ or (C) NeuN+ double-positive cells. The light-gray dashed lines indicate the border between the VZ and SVZ/neuron-enriched zone. The images were acquired using a Zeiss LSM 800 confocal microscope with a 20x objective. Scale bars = 100 µm. Abbreviations: DAPI = 4',6-diamidino-2-phenylindole; dps = days post seeding; GFP = green fluorescent protein; NeuN = neuronal nuclei protein; PAX6 = paired box 6 protein; SVZ = subventricular zone; VZ = ventricular zone. Please click here to view a larger version of this figure.
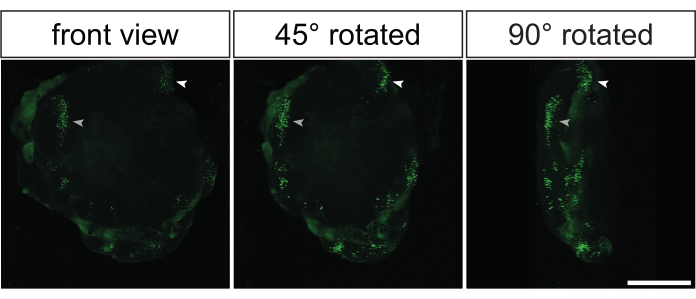
Figure 5: Three-dimensional reconstruction of electroporated images by 3D confocal imaging of electroporated primate cerebral organoids after optical clearing. Frontal (left image), 45° rotated (middle image), and 90° rotated (right image) views of a 3D reconstructed electroporated 32 dps human cerebral organoid 2 days after electroporation with the GFP-expressing plasmid. Prior to imaging, the organoid was optically cleared based on the 2Eci method35. A 3D reconstruction of whole electroporated organoid was generated from 269 optical sections (1-µM thickness each) that are 3.73 µm apart from each other using a Zeiss LSM 800 confocal microscope with a 10x objective. The images were processed for 3D reconstruction using Fiji. Note that the images were taken from the same 3D-reconstructed organoid shown in Video 1. Scale bar = 500 µm. Abbreviation: GFP = green fluorescent protein. Please click here to view a larger version of this figure.
Video 1: A 3D-reconstructed electroporated human cerebral organoid after optical clearing. Video of a 3D-reconstructed electroporated 32 dps human cerebral organoid 2 days after electroporation with the GFP-expressing plasmid. Prior to imaging, the organoid was optically cleared based on the 2Eci method35. A 3D reconstruction of whole electroporated organoid was generated from 269 optical sections (1-µM thickness each) that are 3.73 µm apart from each other using a Zeiss LSM 800 confocal microscope with a 10x objective. The images were processed for 3D reconstruction using Fiji. Note that the video was taken from the same 3D-reconstructed organoid shown in Figure 5. Please click here to download this Video.
| iPSC line | Species | Publication | Culture medium composition | Culture conditions | ||||
| iLonza2.2 | Homo sapiens | Stauske et al., 2020 | 1 µM IWR1 and 0.5 µM CHIR in StemMACS iPS-Brew XF | humified atmosphere of 5% CO2 and 95% air, 37 °C | ||||
| SandraA | Pan troglodytes | Mora-Bermúdez et al., 2016 | mTeSR1 | humified atmosphere of 5% CO2 and 95% air, 37 °C | ||||
| iRh33.1 | Macaca mulatta | Stauske et al., 2020 | 1 µM IWR1 and 0.5 µM CHIR in StemMACS iPS-Brew XF | humified atmosphere of 5% CO2 and 95% air, 37 °C | ||||
| cj_160419_5 | Callithrix jacchus | Petkov et al., 2020 | 3 µM IWR1, 0.3 µM CGP77675, 0.3 µM AZD77675, 0.5 µM CHIR99021, 10 µM Forskolin, 1 ng/mL Activin A, 1 µM OAC1 in StemMACS iPS-Brew XF | humified atmosphere of 5% CO2, 5% O2, and 90% N2, 37 °C | ||||
Table 1: Culture conditions for the primate iPSCs used in this publication. Abbreviation: iPSCs = induced pluripotent stem cells.
| Medium | Composition | ||
| Neural induction medium | 1x N-2 supplement, 1x Glutamine substitute Supplement, 1x MEM Non-Essential Amino Acids Solution, 1 µg/mL Heparin in Dulbecco's Modified Eagle Medium F12 (DMEM/F12) | ||
| Differentiation medium (DM) without Vitamin A | 0.5x B-27 Supplement (minus vitamin A), 0.5x N-2 supplement, 0.5x MEM Non-Essential Amino Acids Solution, 1x Glutamine substitute Supplement, 100 U/mL Penicillin-Streptomycin, 0.00035% 2-Mercaptoethanol, 2.875 ng/mL Insulin in 1:1 DMEM/F12 and Neurobasal Medium | ||
| Differentiation medium (DM) with Vitamin A | 0.5x B-27 Supplement, 0.5x N-2 supplement, 0.5x MEM Non-Essential Amino Acids Solution, 1x Glutamine substitute Supplement, 100 U/mL Penicillin-Streptomycin, 0.00035% 2-Mercaptoethanol, 2.875 ng/mL Insulin in 1:1 DMEM/F12 and Neurobasal Medium | ||
Table 2: Composition of the media used for primate cerebral organoid generation and culture.
| Component | Control electroporation mix | GOI electroporation mix |
| GFP expression plasmid | 500 ng/µL | 500 ng/µL |
| Empty vector | 500 ng/µL | – |
| GOI expression plasmid | – | 500 ng/µL |
| Fast Green | 0.10% | 0.10% |
| in DPBS |
Table 3: Composition of the electroporation mix (separate plasmids approach) for the control and gene of interest. Abbreviation: GOI = gene of interest.
| Antibody | Company | Catalog Number | RRID | Dilution |
| Chicken anti GFP | Aves labs | GFP-1020 | RRID:AB_10000240 | 1:300 |
| Rabbit anti PAX6 | Novus Biologicals | NBP1-89100 | RRID:AB_11013575 | 1:300 |
| Rabbit anti NeuN | Abcam | ab104225 | RRID:AB_10711153 | 1:300 |
| Goat anti chicken Alexa Fluor 488 | Thermo Fisher | A-11039 | RRID:AB_142924 | 1:500 |
| Donkey anti rabbit Alexa Fluor 555 | Thermo Fisher | A-31572 | RRID:AB_162543 | 1:500 |
Table 4: Antibodies used for immunofluorescence staining.
Supplemental Figure S1: VZ/SVZ border determination in electroporated primate cerebral organoids. Double immunofluorescence for PAX6 (magenta) and TUJ1 (yellow) combined with DAPI staining (cyan) of a 32 dps marmoset cerebral organoid 2 days after electroporation with the GFP-expressing plasmid. The immunofluorescence for GFP is not shown. The light-gray dashed lines indicate the border between the VZ and SVZ/neuron-enriched zone. The images were acquired using a Zeiss LSM 800 confocal microscope with a 20x objective. Scale bar = 100 µm. Abbreviations: DAPI = 4',6-diamidino-2-phenylindole; dps = days post seeding; PAX6 = paired box 6 protein; SVZ = subventricular zone; TUJ1 = class III β-tubulin; VZ = ventricular zone. Please click here to download this File.
Supplemental File 1: Petri dish electroporation chamber assembly instructions. Please click here to download this File.
