세포 기능의 분자 염기를 규명하기 위해서는 세포 배양에서 형질전환 DNA의 발현이 필요한 경우가 많습니다. 발현되기 위해서는 전이유전자가 세포의 선택적 막을 뚫고 들어가 핵 1,2에 도달해야 합니다. 따라서 세포의 물리적 장벽을 효과적으로 우회하고 중심 과정을 조작하는 능력은 새로운 생물학적 현상을 밝히기 위해 형질전환을 적용하는 데 필수적입니다. 한 가지 접근법은 바이러스가 외래 DNA를 전달하고 발현하는 고유한 능력을 이용하는 것이다 3,4.
아데노 관련 바이러스(AAV)는 가장 작은 포유류 바이러스 중 하나로, 4.7킬로베이스(kb)의 단일 가닥 DNA 게놈에는 25nm 크기의 60mer 이십면체 캡시드 안에 포장된 rep(복제용)와 cap(캡시드용)이라는 두 개의 유전자가 포함되어 있습니다. rep/cap 유전자는 바이러스 복제, 생산 및 패키징에 필요한 최소 9개의 고유한 단백질을 암호화하는 다중 프로모터, 판독 프레임 및 스플라이스 산물을 가지고 있습니다 5,6. 또한 게놈의 양쪽 말단에는 형질도입 중 DNA 복제, 게놈 패키징 및 다운스트림 처리에 필요한 ITR(Inverted Terminal Repeat)이라는 2차 구조가 포함되어 있습니다 7,8,9,10. ITR은 게놈을 캡시드로 패키징하는 데 필요한 유일한 DNA 요소이므로, AAV는 바이러스 rep/cap 유전자를 연구자가 선택한 조절 요소 및/또는 관심 유전자로 대체하여 전이유전자 전달 목적으로 클로닝할 수 있다6. 그 결과 조작된 벡터 게놈(VG)을 가진 재조합 AAV(rAAV)는 인간 유전자 치료를 위한 클리닉에서 널리 사용되며 성공을 거두었습니다11. 벡터의 과소 평가 된 사용은 실험실에 있습니다. rAAV는 배양된 세포에서 전이유전자 발현을 효율적으로 달성하여 연구자의 실험적 요구를 충족시킬 수 있다12.
rAAV를 생산하는 가장 일반적인 방법은 HEK293 또는 293T 세포에 삼중 플라스미드 transfection을 하는 것입니다(그림 1). 일반적으로 시스 플라스미드(cis plasmid)라고 불리는 첫 번째 플라스미드는 ITR(pAAV)이 측면에 있는 원하는 전이유전자(transgene)를 포함합니다. 용도에 따라 strong promoter 또는 CRISPR 기반 도구와 같은 공통 요소를 가진 cis plasmid를 구입할 수 있습니다. 두 번째는 trans 내에서 제공되는 야생형 AAV rep 및 cap 유전자를 포함하는 pRep/Cap 플라스미드로, cis 플라스미드와 상호 작용하는 조절 및 구조 요소를 발현하는 별도의 non-ITR 함유 플라스미드에 포함되어 있으므로 trans 플라스미드라고 합니다. VG를 물리적으로 둘러싸는 것 외에도 캡시드는 세포 영양성12,13에 영향을 미칩니다. 혈청형 특이적 캡 유전자를 trans 내에서 제공함으로써 연구자들은 주어진 표적 세포에 최적화된 캡시드 혈청형을 선택하여 형질도입 효율을 쉽게 극대화할 수 있습니다. 마지막으로, Dependoparvovirus로서, AAV는 pAdΔF614,15와 같은 제3의 플라스미드에 제공된 아데노바이러스 도우미 유전자에 의해 달성되는 바이러스 프로모터의 rep/cap 발현을 활성화하기 위해 도우미 바이러스를 필요로 합니다. 72시간의 triple-plasmid transfection 후, 벡터는 반복적인 동결/해동 주기를 통해 생산자 세포에서 배양 배지로 방출될 수 있습니다. 그런 다음 전체 플레이트 내용물을 수집하고 원심 분리로 큰 세포 파편을 제거합니다. 생성된 배지 상층액은 다운스트림 형질도입을 위해 준비된 미처리 rAAV 제제입니다.
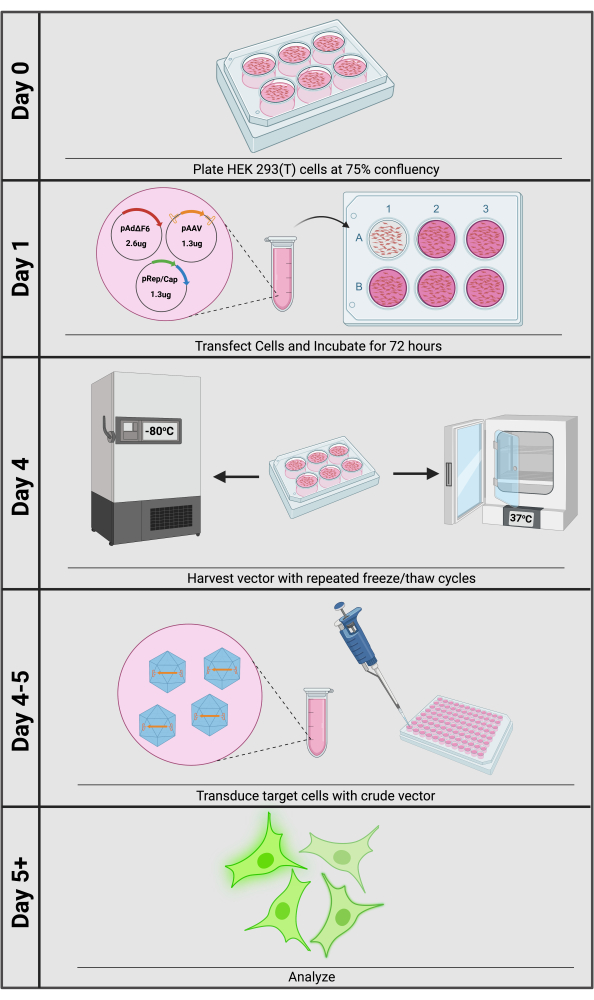
그림 1: 미처리 rAAV 벡터 생산 개요. 원유 rAAV 생산 및 형질도입은 5일 이내에 완료될 수 있습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
rAAV는 전기천공법 또는 화학적/지질-기반 형질주입과 같이 세포 독성, 낮은 효율, 고가의 시약 및 장비와 일반적으로 연관되는 다른 형질주입 방법에 비해 전이유전자 전달에 더 유리할 수 있다16,17. rAAV는 이러한 장애물을 우회하여 독성을 최소화하고 실습 시간을 최소화하면서 강력한 전이유전자 발현을 제공하는 경우가 많습니다. 중요한 것은 rAAV를 생산하고 세포 배양에 적용하는 것이 간단하며 배양 배지에서 벡터를 정제할 필요가 거의 없다는 것입니다(그림 1). 또한, rAAV는 렌티바이러스 전이유전자 전달(Lentiviral transgene delivery)과 달리 VG를 숙주 게놈에 통합하지 않으며, 따라서 삽입 돌연변이 유발(insertional mutagenesis)의 위험을 낮춘다18. 전이유전자 전달을 위해 rAAV를 사용하는 것의 잠재적인 이점에도 불구하고, 한계점을 고려해야 한다. 중요한 것은 ITR을 포함한 전이유전자의 크기가 캡시드의 물리적 제약으로 인해 4.9kb를 초과해서는 안 되며, 이로 인해 연구자가 큰 조절 요소와 전이유전자를 효과적으로 전달할 수 있는 능력이 제한되어서는 안 된다는 것입니다. 또한, rAAV는 비통합 바이러스이기 때문에, 형질도입은 분열하는 세포에서 일시적인 전이유전자 발현을 초래하며, 안정적인 발현을 위해 실용적이지 않을 수 있다. 그러나, 연구자가 원하는 경우 이중 rAAV-전달 Cas9 및 상동성 지향 복구(homology-directed repair, HDR) 템플릿을 사용하는 방법을 사용하여 특정 게놈 유전자좌(genomic loci)에 서열을 안정적으로 삽입할 수 있다19.
클로닝
클로닝 프로토콜은 위에서 사용한 pAAV.CMV.Luc.IRES.EGFP.SV40 플라스미드에 국한되지 않으며 연구자의 실험적 필요에 따라 쉽게 변경할 수 있습니다. 많은 ITR 함유 플라스미드는 온라인에서 쉽게 구입할 수 있습니다. 예를 들어, Cas9 및 sgRNA 클로닝 부위를 모두 포함하는 플라스미드를 사용할 수 있지만 올리고뉴클레오티드 어닐링 및 PNK 처리와 같은 몇 가지 추가 단계가 필요합니다30. 또한, ITR만 있고 내부 조절 요소가 없는 다중 클로닝 사이트(MCS)를 함유하는 플라스미드가 발견될 수 있다31. 다른 플라스미드를 사용하는 경우, 분해에 사용되는 제한 효소(RE)는 일반적으로 이 프로토콜에서 변경해야 할 수 있는 유일한 요소입니다. 그러나 rAAV의 한계는 제한된 화물 용량입니다. 캡시드의 물리적 한계로 인해 벡터 게놈은 ITR을 포함하여 4.9kb를 초과해서는 안 됩니다.
박테리아에서 플라스미드를 분리할 때, 삼중 플라스미드 transfection 또는 transduction 중 세포에 대한 손상을 완화하기 위해 endotoxin-low 또는 -free midiprep 또는 maxiprep kit를 사용하는 것이 중요합니다. miniprep 키트의 플라스미드는 종종 더 높은 불순물, 감소된 농도 및 더 적은 supercoiled DNA를 포함하며, 이 모든 것이 rAAV의 다운스트림 생산에 영향을 미칠 수 있으므로 권장되지 않습니다.
복제 중에 ITR의 구조와 특성을 이해하는 것이 중요합니다. 첫째, ITR을 통해 PCR을 사용하는 것은 매우 어렵습니다. ITR을 통한 PCR 증폭이 필요한 클로닝 설계는 피해야 하며, Gibson 어셈블리 클로닝 기술의 사용을 추가로 제한해야 합니다. 따라서, 제한효소 클로닝은 ITR-함유 플라스미드로의 클로닝에 선호되는 방법이다. 또한, Sanger 염기서열분석을 위한 특정 프라이머는 염기서열분석된 영역에 ITR이 포함된 경우 호환되지 않을 수 있습니다. 대신, 보다 정확한 염기서열 분석 결과를 얻기 위해 ITR에서 벡터 게놈 본체로 염기서열을 분석하는 프라이머를 사용하는 것이 좋습니다. 둘째, ITR은 플라스미드 증폭을 위해 박테리아로 형질전환될 때 결실, 재배열 및 돌연변이가 발생하기 쉽습니다32,33. 이러한 현상을 완화하려면 Stbl3와 같은 재조합 결핍 유능한 박테리아 균주를 사용하고 세포 분열을 늦추기 위해 30°C에서 배양하는 것이 좋습니다. 마지막으로, ITR이 없는 콜로니가 성장 이점을 제공하고 더 클 수 있기 때문에 더 작은 콜로니는 재배열이나 삭제 없이 클론과 일치할 수 있다는 것이 관찰되었습니다. 따라서 작은 식민지를 선택하는 것이 좋습니다.
벡터 제작
rAAV 벡터의 성공적인 생성은 여러 요소의 영향을 받을 수 있습니다. 한 가지 중요한 요소는 transfection에 사용되는 HEK293 또는 293T 세포의 상태입니다. 일반적으로 낮은 통로 수가 이상적인데, 이는 높은 통로 세포가 rAAV 역가를 감소시킬 수 있는 유전형 및 표현형 변이를 나타낼 수 있기 때문입니다. 또한 효과적인 생산을 위해 파종된 세포의 밀도는 75%-90% 밀도여야 합니다. 희소 세포는 벡터를 생성하는 데 사용할 수 있는 세포가 적기 때문에 벡터 수율이 낮고, 과도하게 자란 세포는 효율적으로 transfection되지 않습니다.
시약 로트, 세포 스톡 및 일반적인 실험실 간 변동성 간의 차이는 transfection 효율 및 생산 titer의 차이에 기여합니다. 역가 개선으로 이어질 수 있는 최적화 가능한 인자 중 하나는 transfection 반응에서 plasmid:PEI 비율입니다. 신선한(<1개월) PEI MAX를 사용하는 것이 중요합니다. 1:1의 plasmid:PEI 비율을 시작점으로 사용하는 것이 좋으며, transfection 또는 transduction 효율이 좋지 않은 경우 여러 가지 다른 비율을 테스트합니다. 역가 최적화는 클로닝을 위한 출발 물질로서 본원에 사용된 CMV.Luc.IRES.EGFP 리포터 trangene과 같은 시각적 판독을 갖는 전이유전자를 사용하는 경우에 가장 쉽다. 최적화를 수행하려면 12웰 플레이트를 사용하고 플라스미드 질량과 시약 부피를 2씩 축소하는 프로토콜 단계 3을 따르십시오(최종 플라스미드 질량은 2.6μg). 1:0.75에서 1:3 범위의 비율에 맞게 PEI 부피를 0.25씩 증가시킵니다(그림 6). 각 반응을 15분 후 950μL의 SF 배지로 희석합니다. 편의를 위해, 3중 플라스미드를 포함하는 마스터 믹스를 만들어 PEI를 추가하기 전에 1.5 mL 튜브에 개별적으로 피펫팅할 수 있습니다( 보충 파일 2 참조). 벡터를 채취하고, 관심 세포를 transduction하고, 이미지를 생성합니다. 형질도입 효율(GFP+ 세포의 비율)이 가장 높은 웰은 PEI:DNA의 가장 높은 역가와 최적 비율에 해당합니다.
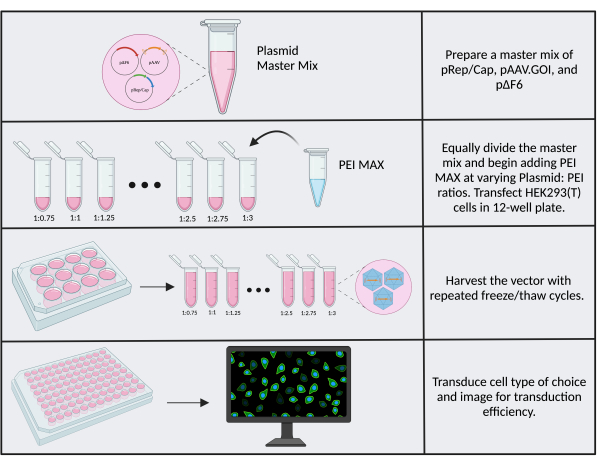
그림 6: PEI 최적화 워크플로우. PEI 최적화에 필요한 단계 개략도. 플라스미드의 여러 비율: 최적의 비율을 결정하기 위해 PEI를 테스트합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
수확 및 역가 고려 사항
rAAV 벡터를 채취하는 데 사용되는 동결/해동 기술은 정제된 용해물을 직접 사용하여 배양된 세포를 transduction하는 것과 호환되는 방식으로 HEK293 세포를 효과적으로 용해합니다. AAV1, AAV8 및 AAV9와 같은 특정 rAAV 혈청형은 벡터 생산 동안 세포로부터 방출되고, 동결/해동 주기 없이 배양된 세포 배지로부터 수거할 수 있다34. 여기에 설명된 분석법은 일반적으로 AAV2 캡시드를 사용할 때 1 x 10 10 VG/mL 정도, AAV8 사용 시 1 x10 11 VG/mL 정도의 역가를 산출합니다. 세제 또는 기타 화학 기반 용해를 통해 더 높은 역가를 얻을 수 있지만, 이는 다운스트림 사용 시 세포에 유해하며 용해물에서 rAAV를 추가로 정제해야 합니다. 낮은 역가는 연구자가 미정제 제제가 연구 요구에 적합한지 여부를 결정할 때 고려해야 하는 한 가지 절충안이지만, 여기에 설명된 방법으로 생성된 약간 낮은 역가는 많은 세포 유형을 매우 잘 transduction할 수 있습니다(대표적인 결과 참조). transfection 효율 및 세포 건강 외에도 벡터 역가는 rAAV 생산 중에 사용되는 캡시드와 VG35 내 전이유전자의 크기 및 서열에 따라 달라집니다.
미정제 벡터 제제를 수확할 때 triple-plasmid transfection 중에 사용된 plasmid DNA가 존재할 수 있으며, 드물지만 transduction 중 downstream transfection을 초래합니다. 더욱이, 포장되지 않은 VG는 캡시드의 외부에 결합할 수 있고, 벌거벗은 및 외래 단일가닥 DNA에 대한 선천성 면역 반응을 불러일으킬 수 있다36,37. 따라서 민감한 세포 유형에서는 DNase를 분해하고 정제하여 포장되지 않은 VG와 플라스미드를 제거해야 할 수 있습니다.
조잡한 제제의 역가를 계산하고자 하는 경우, qPCR을 수행하여 DNase 내성 입자(DRP) 내에 포장된 VG의 수를 정량화할 수 있습니다. 간단히 말해서, 소량의 미정제 제제를 DNase 분해하여 플라스미드 DNA, 오염 핵산 또는 부분적으로 포장된 VG를 제거합니다. 그런 다음 샘플을 qPCR에 적용하고 DRP 내부의 보호된 VG를 정량화하여 미처리 제제(38)의 mL당 벡터 게놈 단위를 갖는 역가를 생성합니다. 캡시드 역가를 정량화하는 ELISA 기반 분석을 사용하여 벡터 적정을 수행하는 것은 권장되지 않습니다. 야생형 AAV 바이러스와 비교했을 때, rAAV는 비어 있거나 부분적으로 포장된 캡시드의 비율이 높다39. ELISA는 게놈 함량에 관계없이 모든 캡시드를 정량화하고 포장된 VG가 필요한 제제에 존재하는 형질도입 가능한 단위를 과대평가합니다.
Transduction 고려 사항
많은 요인이 rAAV transduction에 영향을 미치며 새로운 실험에 대해 적절한 고려가 이루어져야 합니다. 전이유전자 발현을 유도하는 프로모터에 따라, 발현 개시는 transduction 후 4시간(hpt)에 발생할 수 있으며, 피크 발현은 일반적으로 48 hpt에 의해 달성됩니다. 세포의 초기 파종부터 실험 종점까지의 기간을 염두에 두는 것이 중요합니다. 이것은 세포의 시작 밀도를 추정하고 실험이 끝날 때까지 과도하게 자라지 않도록 하기 위한 것입니다. 세포가 과밀화되면 스트레스 반응으로 인해 세포 행동이 변경되어 실험 결과를 혼란스럽게 할 수 있습니다. U2-OS와 같은 일부 세포 유형은 과증식/접촉 억제를 잘 견딜 수 있습니다. 또한 이 생산 프로토콜의 산물인 무혈청 컨디셔닝 배지에서 장기간(48h+)을 견딜 수 있습니다. 그러나, 민감한 세포 유형은 형질도입 중 건강을 유지하기 위해 특수 성장 배지로 미처리 제제의 혈청 추가 또는 희석이 필요할 수 있습니다. 혈청 함유 배지 사용으로 인한 형질도입 효율이 약간 감소하는 것은 세포 건강에 대한 잠재적인 절충안이며 연구자는 이를 고려해야 합니다.
일반적으로 빠르게 분열하는 세포의 경우 약 50%의 시작 밀도가 48hpt로 종료되는 응용 분야에 최적입니다. 그러나 밀도는 실험의 필요에 따라 적절하게 조정할 수 있습니다. transduction 효율 감소로 인해 75% 이상의 밀도를 초과하는 단층 유형의 불멸화된 세포주를 transduction하는 것은 권장되지 않습니다. 대부분의 배양된 세포 유형은 조잡한 rAAV 제제로 하룻밤 동안 배양한 후 아침에 신선한 혈청 함유 배지로 교체한 후 성공적으로 transduction되고 건강합니다.
캡시드 혈청형은 표적 세포를 transduction하기 위해 rAAV를 생산할 때 고려해야 할 중요한 요소인데, 이는 capsid가 세포 tropism 및 후속 transgene 발현의 주요 결정 인자이기 때문이다13. AAV2는 많은 유형의 배양 세포를 효과적으로 transduction할 수 있는 능력으로 인해 널리 사용되는 혈청형이다12. AAV2의 이러한 특성은 AAV2에 대한 1차 부착 인자 역할을 하는 헤파린 설페이트 프로테오글리칸(HSPG) 및 배양된 세포에서 적응에서 성장에 이르기까지 높은 수준의 HSPG에 기인할 수 있다(40). AAV9와 같은 다른 캡시드는 광범위한 세포 유형을 형질도입하는데 덜 효과적이며, 이러한 설정에서 발현되지 않는 의존 부착 인자에 의해 설명될 수 있다(41). 따라서 원하는 표적 세포가 이전에 문헌에서 rAAV로 테스트되지 않은 경우 배양된 세포에서 AAV2를 1순위 캡시드로 권장합니다.
미처리 벡터 제제의 주요 한계는 동물 모델을 형질도입하는 데 부적절하다는 점에 유의하십시오. 생체 내 연구에서는 정제 및 품질 평가를 위한 준비가 필요합니다.
전이유전자 발현 및 잠재적 통합 고려사항
rAAV는 전이유전자의 영구적인 발현을 확실하게 초래하지 않습니다. 시간이 지남에 따라, VG는 침묵될 수 있고, 형질전환 발현은 몇몇 구절42에 따라 중단될 수 있다. 또한, VG의 대다수는 일시성체로 남아 있으며, rAAV는 야생형 바이러스 용원 감염에서와 같이 숙주 게놈으로의 빈번한 통합을 매개하거나 VG의 복제를 촉진하는 바이러스 Rep 단백질을 포함하지 않는다43. 결과적으로, 형질도입된 세포의 에피솜은 결국 분열을 통해 딸 세포 사이에서 희석됩니다.
기초 수준 통합은 전달된 모든 형질전환 DNA 물질에 대한 가능성입니다. 그러나, ITR-함유 VG들은 더 높은 주파수(44)에서 통합되는 경향이 있다. 그러므로, 전이유전자의 영구적인 발현은 세포의 작은 부분집합에서 관찰될 수 있다. 사용자는 특히 Cas9과 같은 DNA 절단 효소를 전달하기 위해 rAAV를 사용할 때 이중 가닥 절단이 통합 및 영구 발현의 훨씬 더 큰 빈도를 초래할 수 있기 때문에 이러한 가능성을 고려해야 합니다(45). 이로 인해 rAAV는 내인성 태깅 또는 유전자 추가를 위한 상동성 지향 복구 템플릿을 전달하기 위한 좋은 후보이지만 Cas9 삽입 가능성은19,46으로 간주되어야 합니다.
