Трехмерная биопечать кокультур нейронов и астроцитов человека, полученных из ИПСК человека, для скрининговых применений
Summary
Здесь мы представляем протокол для получения 3D-биопечатных кокультур нейронов и астроцитов, полученных из ИПСК. Эта модель кокультуры, созданная в гидрогелевом каркасе в 96- или 384-луночном формате, демонстрирует высокую постпечатную жизнеспособность и рост нейрита в течение 7 дней и показывает экспрессию маркеров зрелости для обоих типов клеток.
Abstract
Для того, чтобы клеточная модель была жизнеспособной для скрининга лекарственных препаратов, система должна соответствовать требованиям к пропускной способности и однородности, а также иметь эффективное время разработки. Однако многие опубликованные 3D-модели не удовлетворяют этим критериям. Таким образом, это ограничивает их полезность в ранних приложениях для разработки лекарств. Трехмерная (3D) биопечать — это новая технология, которая может быть применена для разработки 3D-моделей для ускорения разработки, повышения стандартизации и увеличения производительности. В данной статье мы представляем протокол для разработки 3D-биопечатных моделей кокультур глутаматергических нейронов и астроцитов, полученных из индуцированных плюрипотентных стволовых клеток человека (ИПСК). Эти кокультуры встроены в гидрогелевую матрицу, состоящую из биоактивных пептидов, белков полноразмерного внеклеточного матрикса (ECM) и имеют физиологическую жесткость 1,1 кПа. Модель может быть быстро установлена в 96-луночном и 384-луночном форматах, а средняя послепечатная жизнеспособность составляет 72%. Показано, что соотношение астроцитов и нейронов в этой модели составляет 1:1,5, что находится в пределах физиологического диапазона для человеческого мозга. Эти 3D-биопечатные клеточные популяции также показывают экспрессию маркеров типа зрелых нейральных клеток и рост проекций нейритов и астроцитов в течение 7 дней после культивирования. В результате эта модель пригодна для анализа с использованием клеточных красителей и методов иммуноокрашивания наряду с анализом роста нейритов. Возможность создавать эти физиологически репрезентативные модели в большом масштабе делает их идеальными для использования в скрининговых анализах со средней и высокой пропускной способностью для целей нейробиологии.
Introduction
Исследования заболеваний центральной нервной системы (ЦНС) в индустрии разработки лекарств расширяются1. Тем не менее, многие распространенные заболевания ЦНС, такие как эпилепсия, шизофрения и болезнь Альцгеймера, до сих пор не поддаются лечению 2,3,4. Отсутствие эффективных методов лечения заболеваний ЦНС может, по крайней мере частично, объяснить отсутствием точныхмоделей головного мозга in vitro. Это привело к трансляционному разрыву между текущими моделями in vitro и данными in vivo и, как следствие, к узкому месту в исследовательских усилиях.
В связи с этим трансляционным разрывом в последние годы наблюдается значительный рост разработки новых 3D-моделей клеток, включая нейронные органоиды, нейросфероидыи модели на основе скаффолдов. 3D-структура этих моделей помогает рекапитулировать нейронное микроокружение, включая биомеханические стрессы, межклеточные контакты и внеклеточный матрикс мозга (ECM)7. ВКМ головного мозга является динамическим элементом нейрофизиологии, который занимает пространство между типами нервных клеток, включая нейроны, астроциты, олигодендроциты и нервно-сосудистый блок7. Было показано, что рекапитуляция ВКМ мозга влияет на морфологию нейронов и возбуждение нейронов, и многие сложные 3D-модели мозга продемонстрировали отложение белков ВКМ нервными клеткамитипов 8,9,10,11. Модели, основанные на скаффолдах, состоят из зрелых нейронных кокультур, подвешенных в пористой синтетической или биологической гидрогелевой матрице, которая представляет собой ECM12 мозга. В отличие от органоидных и сфероидных систем, 3D-модели на основе скаффолдов позволяют настраивать присутствующие белки ECM и имеют дополнительное преимущество в виде настраиваемости жесткости гидрогеля для имитации биомеханических напряжений13,14.
Несмотря на то, что подавляющее большинство 3D-нейронных моделей демонстрируют повышенную рекапитуляцию микроокружения мозга, не все модели хорошо подходят дляреализации приложений для разработки лекарств. Для того чтобы 3D-модель могла быть внедрена в промышленные процессы, система должна соответствовать требованиям к пропускной способности для применения в области скрининга и иметь относительно короткое время разработки16. 3D-биопечать — это новая технология, которая предлагает потенциал для создания нейронных моделей на основе 3D-каркасов с быстрым временем разработки, повышенной производительностью и более высоким уровнем точности контроля, наряду с устранением вариативности, вызваннойчеловеческой ошибкой. Этот протокол представляет собой 3D-модель кокультуры глутаматергических нейронов и астроцитов человека, полученных из ИПСК, в гидрогелевом каркасе. Этот гидрогелевый каркас содержит физиологически репрезентативные биоактивные пептиды (RGD, IKVAV, YIGSR) и белки ECM в миметической биомеханической жесткости. Эти полноразмерные белки ECM включают полноразмерный ламинин-211 и гиалуроновую кислоту, в изобилии присутствующие в коре головного мозга человека, с жесткостью 1,1 кПа, что соответствует измерениям in vivo 18. Эта модель разработана с практичностью для разработки лекарств и создается с помощью 3D-биопринтера в формате 96-луночной или 384-луночной пластины, подходящей для скринингового анализа с использованием методов визуализации с клеточными красителями и антителами, а также анализов роста нейритов. Клетки демонстрируют экспрессию маркеров типа нейральных клеток и рост нейритовых и астроцитарных проекций в течение 7 дней после культивирования. Таким образом, в этом протоколе будет представлена методология разработки высокопроизводительной 3D-модели нейронной кокультуры для использования в приложениях для разработки лекарств.
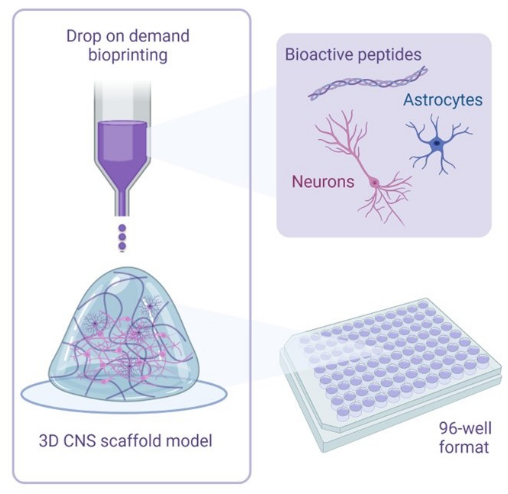
Рисунок 1: Иллюстративный обзор методологии, используемой для 3D-биопечати кокультур. Нейроны и астроциты, полученные из ИПСК человека, комбинируются с растворами активаторов и биочернил, содержащими биоактивные пептиды, и биопечатаются на гидрогелевые каркасы в 96-луночном или 384-луночном форматах с использованием технологии биопечати «капля по требованию». Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
Protocol
Representative Results
Discussion
Потребность в точных моделях ЦНС никогда не была столь высокой, а ограничения двумерных (2D) традиционных моделей клеточных культур привели к появлению впоследние годы поколения сложных моделей ЦНС. Однако многие сложные 3D-модели, представляющие взаимодействия между типами нервных клеток и ECM, имеют ограничения, которые препятствуют применению этих моделей в промышленных процессах 6,20,21. В рамках этого протокола мы разрабатываем 3D-модель кокультуры нейронов и астроцитов, полученных из ИПСК человека, которая направлена на устранение некоторых из этих ограничений с помощью технологии 3D-биопечати для создания биоактивного гидрогелевого каркаса в 96-луночном и 384-луночном форматах.
Методология разработки этих моделей была упрощена благодаря программному обеспечению для проектирования карт пластин, автоматически генерируемым протоколам печати и управляемому процессу печати с биопринтера. Тем не менее, из-за чувствительного характера чувствительных типов клеток, полученных из ИПСК, используемых в этом протоколе, следует соблюдать следующие критические меры по размораживанию и культивированию. Во-первых, включение ингибитора ROCK (ROCKi) имеет ряд преимуществ на протяжении всего процесса биопечати и во время раннего культивирования. Оттаивание клеток является критической точкой, в которой нейроны могут испытывать стрессовую реакцию, а неправильные протоколы размораживания могут снизить шансы навыживание. Как правило, рекомендуется как можно эффективнее размораживать клетки, добавлять среду и поднимать клетки до температуры инкубатора23. Однако во время процесса биопечати, описанного в этом протоколе, необходимо, чтобы нейроны и астроциты ресуспендировали в растворе активатора, а не в среде, и клетки не поднимались выше комнатной температуры до конца печатного тиража (до 30 минут после оттаивания). Таким образом, добавление ROCKi в среду сразу после размораживания и включение его в течение двух стадий центрифугирования (этапы 2.1–2.7 и 1.3.15-1.3.20) является обязательным для ингибирования путей клеточного стресса, что приведет к снижению уровня жизнеспособности24. Кроме того, было показано, что ROCKi способствует росту нейритов и улучшает созревание нейронов25. Таким образом, прием добавок ROCKi продолжается в течение 48 ч после биопечати. Тем не менее, необходимо удалить добавку ROCKi через 48 ч, чтобы обеспечить полное смывание при последующей смене среды перед использованием клеток для анализа.
Следующим этапом, требующим критического внимания, является добавление и смена носителей после печати (шаги 2.8-2.13). Биопечатный гидрогелевый каркас имеет эквивалентную биомеханическую жесткость всего 1,1 кПа, что эквивалентно серому веществу. Как описано на шаге 2.10, во время добавления и аспирации среды очень важно осторожно вводить пипетку в стенку лунки, чтобы предотвратить нарушение. Это особенно актуально для 384-луночных планшетов, где уровень геля занимает большую долю от общего объема лунки. Этот метод также следует использовать в 2D-контрольных скважинах для предотвращения подъема краев клеток и сдвига нейритовых выростов. Авторы также хотели бы подчеркнуть важность стерильной техники в биопринтере, к которой следует относиться с такой же осторожностью, как и к шкафу биобезопасности, используемому для клеточных культур, полученных из ИПСК. Это включает в себя стерильную фильтрацию 70% EtOH и dH2O, используемую в процедурах «зеленого света» и печати, сохранение крышек на картриджах и пластинах при перемещении рук внутрь и из биопринтера, а также обеззараживание поверхностей внутри биопринтера салфетками с 70% этанолом до и после печати.
Биопечатный гидрогелевый каркас, сформированный из биочернил и растворов активаторов, выбранный для разработки этой модели, выбран из ряда биочернил и растворов-активаторов, разработанных Inventia Life Science для использования в биопринтере RASTRUM. Ламинин и гиалуроновая кислота были идентифицированы как молекулы, имеющие отношение к созреванию нейронов, полученных из ИПСК, из-за их роли в аксональном направлении, образовании синапсов и формировании перинейрональной сети26,27. Кроме того, была выбрана биомеханическая жесткость 1,1 кПа, поскольку было показано, что гидрогели с более низкой плотностью обеспечивают лучший рост нейритов из нейронов12. Если в протокол вносятся изменения с использованием нейронов и астроцитов, которые были дифференцированы собственными силами или от другого коммерческого поставщика, рекомендуется провести тест выбора матрицы для определения наиболее благоприятного гидрогелевого каркаса15. Кроме того, может потребоваться оптимизация плотности клеток, если вносятся изменения в источники клеток для обеспечения оптимальной жизнеспособности и предотвращения переполнения гидрогеля. По всем вопросам модификации и устранения неполадок, связанных с работой биопринтера, авторы рекомендуют обращаться к производителям и ссылаться на протоколы производителей.
ЦНС содержит широкий спектр подтипов нейронов и глиальных клеток, все из которых существуют в различных нишах мозга и играют специфическую роль, внося свой вклад в нейронную функцию28. В контексте этого широкого охвата эта модель представляет только два наиболее распространенных типа клеток (астроциты и возбуждающие глутаматергические нейроны). Важные типы клеток, такие как микроглия, олигодендроциты и эндотелиальные клетки, формирующие гематоэнцефалический барьер, исключены из этой системы. Включение микроглии может иметь значение при изучении нейроиммунных взаимодействий, а олигодендроциты могут представлять интерес при заболеваниях, влияющих на центральную миелинизацию. В дополнение к своей роли в патологии, такие клетки, как эндотелиальные клетки, образующие гематоэнцефалический барьер, выделяют ферменты, метаболизирующие лекарственные средства, что может повлиять на использование этой модели для фармакокинетических анализов29. Дальнейшим ограничением модели может быть соотношение астроцитов и нейронов; Соотношение астроцитов и нейронов сильно варьируется между областями мозга, с рекомендуемыми значениями от 1:1 до 1:330,31. Эта модель имеет приблизительное соотношение астроцитов к нейронам 1:1,5; Таким образом, эта модель может не иметь отношения к моделированию областей мозга, где астроцитов больше, например, в областях белого вещества30.
В последние годы были опубликованы и другие протоколы для разработки 3D-моделей кокультур с биопечатью. В публикации Sullivan et al., 2021, была представлена 3D-биопечатная нейронная модель с использованием нейральных клеток-предшественников, полученных из ИПСК, которая демонстрирует высокую постпечатную жизнеспособность и усиление функции нейронов по сравнению с 2D-культурами32. Однако в этом протоколе нейральные клетки-предшественники использовались в качестве клеточного источника и поддерживались в культуре в течение 4 недель. В этом протоколе использовались коммерчески доступные глутаматергические нейроны и астроциты, полученные из ИПСК. Это позволяет создать 3D-сеть совместно культивируемых клеток всего за 7 дней; Как показал анализ роста нейритов, рост нейрита начинается в течение 24 ч и продолжается линейно в течение 156 ч, в течение которых наблюдался рост клеток. Быстрое установление этих сетей может быть частично связано с использованием глутаматергических нейронов, которые используют оптимизированную индуцируемую доксициклином экспрессию гена NGN2, которая показывает экспрессию маркеров зрелого подтипа нейронов в течение 7 дней, даже в 2D-культуре33. Сокращение этого периода роста с помощью этого метода имеет важное значение для внедрения моделей в биофармацевтической промышленности, поскольку разработка тестов требует быстрого оборота и разработки клеточных моделей15.
В заключение, эта модель демонстрирует потенциал для 3D-модели нейронов и астроцитов, которая быстро и удобно устанавливается для целей скрининга. В будущем этот тип модели может быть применен для разработки лекарств для различных заболеваний ЦНС, с возможностью расширения для различных заболеваний с использованием линий ИПСК пациента или отредактированного гена. Кроме того, использование индуцируемых доксициклинами NGN2 экспрессии NGN2 на основе глутаматергических нейронов позволяет клеткам достигать зрелости за меньшее время, что может быть использовано для разработки моделей старения мозга для исследований нейродегенерации. Эта система также может быть расширена за счет использования дополнительных типов клеток в кокультуре, включая микроглию и олигодендроциты.
Declarações
The authors have nothing to disclose.
Acknowledgements
Авторы выражают благодарность Алексу Фолькерлингу, Мартину Энгелю и Рэйчел Блич за помощь в разработке протокола и отзывы о рукописи.
Materials
| 2-mercaptoethanol | Thermofisher | 31350010 | |
| 384-well plate | PerkinElmer | 6057300 | |
| 96-well plate | PerkinElmer | 6055300 | |
| Activator fluid F299 | Inventia Life Science | N/A | |
| Activator fluid F3 | Inventia Life Science | N/A | |
| B27 (50x) minus Vit A | Thermofisher | 12587010 | |
| Bioink fluid F261 | Inventia Life Science | N/A | |
| Bioink fluid F32 | Inventia Life Science | N/A | |
| Doxycycline hyclate | Sigma Aldrich | D5207 | |
| GlutaMAX (100x) | Thermofisher | 35050061 | |
| Goat anti-mouse IgG H&L Alexa Fluor 647 | Abcam | ab150115 | |
| Goat anti-rabbit IgG H&L Alexa Fluor 488 | Abcam | ab150077 | |
| Hoechst | Abcam | ab228551 | |
| Human BDNF Recombinant Protein | Thermofisher | PHC7074 | |
| Human NT3 Recombinant Protein | Thermofisher | PHC7036 | |
| iCell Astrocytes | Fujifilm CDI | 1434 | |
| INCell Analyser 6500HS | Molecular Devices | N/A | high content imaging system |
| Incucyte S3 | Sartorius | N/A | |
| ioGlutamatergic Neurons (Large vial) | Bit.bio | e001 | |
| Laminin (1 mg/mL) | Sigma Aldrich | L2020 | |
| Live/dead kit (Calcein-AM, Ethidium homo-dimer-1) | Invitrogen | L3224 | |
| Mouse anti-BIII tubulin NL637 conjugated | R&D systems | SC024 | |
| Neurobasal media | Thermofisher | 21103049 | |
| Normal Donkey Serum | Abcam | ab7475 | |
| NucBlue Live (Hoechst 33342) | Thermofisher | R37605 | |
| Opti-MEM | Thermofisher | 11058021 | |
| Paraformaldehyde | Sigma Aldrich | P6148 | |
| PEI 50% in H2O | Sigma Aldrich | 181978 | |
| Pierce Borate Buffer 20x | Thermofisher | 28341 | |
| Prism | GraphPad | Data analysis software | |
| Rabbit anti-ionotropic glutamatre receptor 2 (GluR2) | Abcam | ab206293 | |
| RASTRUM(TM) Bioprinter | Inventia Life Science | N/A | Bioprinter |
| RASTRUM(TM) Bioprinter Cartridges | Inventia Life Science | N/A | Bioprinter Cartridges |
| RASTRUM(TM) Targeting plate | Inventia Life Science | N/A | Targeting plate |
| Rho kinase (ROCK) inhibitor | Abcam | ab120129 | |
| Sheep anti-GFAP NL493 conjugated | R&D systems | SC024 | |
| Signals Image Artist | PerkinElmer | N/A | Image analysis platform |
| Triton X-100 | Thermofisher | HFH10 | |
| Zeiss Axio Observer | Zeiss | N/A | Inverted microscope platform |
Referências
- Jung, Y. L., Hwang, J., Yoo, H. S. Disease burden metrics the innovations of leading pharmaceutical companies: a global and regional comparative study. Globalization and Health. 16 (1), 80-80 (2020).
- Potkin, S. G., et al. The neurobiology of treatment-resistant schizophrenia: paths to antipsychotic resistance and a roadmap for future research. npj Schizophrenia. 6, 1 (2020).
- Zahra, W., et al., Keswani, C., et al. The Global Economic Impact of Neurodegenerative Diseases: Opportunities and Challenges. Bioeconomy for Sustainable Development. , (2019).
- Perucca, E. The pharmacological treatment of epilepsy: recent advances and future perspectives. Acta Epileptologica. 3 (1), 22 (2021).
- Nikolakopoulou, P., et al. Recent progress in translational engineered in vitro models of the central nervous system. Brain. 143 (11), 3181-3213 (2020).
- Whitehouse, C., Corbett, N., Brownlees, J. 3D models of neurodegeneration: implementation in drug discovery. Trends in Pharmacological Sciences. 44 (4), 208-221 (2023).
- Rauti, R., Renous, N., Maoz, B. M. Mimicking the brain extracellular matrix in vitro: A review of current methodologies and challenges. Israel Journal of Chemistry. 60 (12), 1141-1151 (2020).
- Fawcett, J. W., Oohashi, T., Pizzorusso, T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nature Reviews Neuroscience. 20 (8), 451-465 (2019).
- Lam, D., et al. Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array. Scientific Reports. 9, 4159 (2019).
- Roll, L., Lessmann, K., Brüstle, O., Faissner, A. Cerebral organoids maintain the expression of neural stem cell-associated glycoepitopes and extracellular matrix. Cells. 11 (5), 760 (2022).
- Yan, Y., Bejoy, J., Marzano, M., Li, Y. The use of pluripotent stem cell-derived organoids to study extracellular matrix development during neural degeneration. Cells. 8 (3), 242 (2019).
- Ma, L., et al. 3D bioprinted hyaluronic acid-based cell-laden scaffold for brain microenvironment simulation. Bio-Design and Manufacturing. 3 (3), 164-174 (2020).
- Liaw, C. -. Y., Ji, S., Guvendiren, M. Engineering 3D hydrogels for personalized in vitro human tissue models. Advanced Healthcare Materials. 7 (4), 1701165 (2018).
- Ma, J., Huang, C. Composition and mechanism of three-dimensional hydrogel system in regulating stem cell fate. Tissue Engineering Part B: Reviews. 26 (6), 498-518 (2020).
- Belfiore, L., et al. Generation and analysis of 3D cell culture models for drug discovery. European Journal of Pharmaceutical Sciences. 163, 105876 (2021).
- Langhans, S. A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Frontiers in Pharmacology. 9, 6 (2018).
- Engel, M., Belfiore, L., Aghaei, B., Sutija, M. Enabling high throughput drug discovery in 3D cell cultures through a novel bioprinting workflow. SLAS Technology. 27 (1), 32-38 (2022).
- Takamura, T., et al. Influence of age on global and regional brain stiffness in young and middle-aged adults. Journal of Magnetic Resonance Imaging. 51 (3), 727-733 (2020).
- Slanzi, A., Iannoto, G., Rossi, B., Zenaro, E., Constantin, G. In vitro models of neurodegenerative diseases. Frontiers in Cell and Developmental Biology. 8, 328 (2020).
- de Souza, N. Organoid variability examined. Nature Methods. 14 (7), 655-655 (2017).
- Hernández, D., et al. Culture variabilities of human iPSC-derived cerebral organoids are a major issue for the modelling of phenotypes observed in Alzheimer’s disease. Stem Cell Review and Reports. 18 (2), 718-731 (2022).
- Li, R., et al. Differentiation of human iPS cells into sensory neurons exhibits developmental stage-specific cryopreservation challenges. Frontiers in Cell and Developmental Biology. 9, 796960 (2021).
- Nishiyama, Y., et al. Safe and efficient method for cryopreservation of human induced pluripotent stem cell-derived neural stem and progenitor cells by a programmed freezer with a magnetic field. Neuroscience Research. 107, 20-29 (2016).
- Uhrig, M., Ezquer, F., Ezquer, M. Improving cell recovery: Freezing and thawing optimization of induced pluripotent stem cells. Cells. 11 (5), 799 (2022).
- Harbom, L. J., et al. The effect of rho kinase inhibition on morphological and electrophysiological maturity in iPSC-derived neurons. Cell and Tissue Research. 375 (3), 641-654 (2019).
- Koh, H. S., Yong, T., Chan, C. K., Ramakrishna, S. Enhancement of neurite outgrowth using nano-structured scaffolds coupled with laminin. Biomaterials. 29 (26), 3574-3582 (2008).
- Tarus, D., et al. Design of hyaluronic acid hydrogels to promote neurite outgrowth in three dimensions. ACS Applied Materials & Interfaces. 8 (38), 25051-25059 (2016).
- Brain Initiative Cell Census Network (BICCN). Initiative Cell Census Network (BICCN). A multimodal cell census and atlas of the mammalian primary motor cortex. Nature. 598 (7879), 86-102 (2021).
- Dauchy, S., et al. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochemical Pharmacology. 77 (5), 897-909 (2009).
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia. 62 (9), 1377-1391 (2014).
- von Bartheld, C. S., Bahney, J., Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. The Journal of Comparative Neurology. 524 (18), 3865-3895 (2016).
- Sullivan, M. A., et al. 3D bioprinting of stem cell-derived central nervous system cells enables astrocyte growth, vasculogenesis and enhances neural differentiation/function. bioRxiv. , (2022).
- Pawlowski, M., et al. Inducible and deterministic forward programming of human pluripotent stem cells into neurons, skeletal myocytes, and oligodendrocytes. Stem Cell Reports. 8 (4), 803-812 (2017).
