转录组由样品中所有基因的表达组成,可通过微阵列和 RNA-seq1 等高通量技术进行分析。数据集中一个基因的表达水平称为转录组学特征,表型组和对照组之间转录组特征的差异表示将该基因定义为该表型的生物标志物 2,3。转录组生物标志物在疾病诊断4、生物学机制5和生存分析6、7等研究中得到广泛应用。
健康组织中的基因活性模式携带有关生命的重要信息8,9。这些模式提供了宝贵的见解,并作为理解良性疾病10,11和致命疾病12的复杂发展轨迹的理想参考。基因相互作用,转录组代表其复杂相互作用后的最终表达水平。这种模式被表述为转录调控网络13和代谢网络14等。信使RNA(mRNA)的表达可以通过转录因子(TFs)和长基因间非编码RNA(lincRNA)进行转录调控15,16,17。传统的差异表达分析忽略了这种复杂的基因相互作用,并假设特征间独立性18,19。
图神经网络 (GNN) 的最新进展表明,在从基于 OMIC 的数据中提取重要信息用于癌症研究20 方面具有非凡的潜力,例如,识别共表达模块21。GNNs的先天能力使它们成为模拟基因之间错综复杂的关系和依赖关系的理想选择22,23。
生物医学研究通常侧重于准确预测对照组的表型。这些任务通常表述为二元分类24,25,26。在这里,两个类标签通常编码为 1 和 0、true 和 false,甚至正负27。
本研究旨在提供一种易于使用的协议,用于基于预训练的图注意力网络 (GAT) 参考模型生成转录组数据集的转录调控 (mqTrans) 视图。先前发表的工作26 中的多任务 GAT 框架用于将转录组学特征转换为 mqTrans 特征。使用来自加州大学圣克鲁斯分校 (UCSC) Xena 平台28 的大型健康转录组数据集对参考模型 (HealthModel) 进行预训练,该模型定量测量了从调节因子(TF 和 lincRNA)到靶 mRNA 的转录调控。生成的 mqTrans 视图可用于构建预测模型和检测暗生物标志物。该协议利用来自癌症基因组图谱(TCGA)数据库29 的结肠腺癌(COAD)患者数据集作为说明性示例。在这种情况下,I 期或 II 期的患者被归类为阴性样本,而 III 期或 IV 期的患者被视为阳性样本。还比较了 26 种 TCGA 癌症类型中深色和传统生物标志物的分布。
HealthModel 管道的说明
该协议中采用的方法基于先前发布的框架26,如 图 1 所示。首先,用户需要准备输入数据集,将其输入到建议的 HealthModel 管道中,并获取 mqTrans 特征。详细的数据制备说明在协议部分的第 2 节中提供。随后,用户可以选择将 mqTrans 特征与原始转录组特征相结合,或者仅继续使用生成的 mqTrans 特征。然后,生成的数据集将进行特征选择过程,用户可以灵活地在 k 倍交叉验证中选择他们的首选 k 值进行分类。该协议中使用的主要评估指标是准确性。
HealthModel26 将转录组学特征分为三个不同的组:TF(转录因子)、lincRNA(长基因间非编码 RNA)和 mRNA(信使 RNA)。TF 特征是根据人类蛋白质图谱30,31 中可用的注释定义的。这项工作利用了 GTEx 数据集32 中 lincRNA 的注释。属于 KEGG 数据库33 中第三级通路的基因被认为是 mRNA 特征。值得注意的是,如果 mRNA 特征表现出 TRRUST 数据库34 中记录的靶基因的调控作用,则将其重新分类为 TF 类。
该协议还手动生成调节因子 (regulatory_geneIDs.csv) 和靶标 mRNA (target_geneIDs.csv) 的基因 ID 的两个示例文件。调控特征(TFs和lincRNAs)之间的成对距离矩阵由Pearson相关系数计算,并通过流行的工具加权基因共表达网络分析(WGCNA)36 (adjacent_matrix.csv)进行聚类。用户可以直接将 HealthModel 管道与这些示例配置文件一起使用,以生成转录组数据集的 mqTrans 视图。
HealthModel的技术细节
HealthModel 将 TF 和 lincRNA 之间的复杂关系表示为图形,输入特征用作 V 表示的顶点,顶点间边矩阵指定为 E。每个样品都具有 K 调控特征,符号为 VK×1。具体来说,数据集包含 425 个 TF 和 375 个 lincRNA,样本维数为 K = 425 + 375 = 800。为了建立边矩阵 E,这项工作使用了流行的工具 WGCNA35。连接两个顶 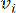 点的成对权重表示为 和
点的成对权重表示为 和 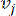 ,由 Pearson 相关系数确定。基因调控网络表现出无标度拓扑结构36,其特征在于存在具有关键功能作用的枢纽基因。我们使用拓扑重叠度量 (TOM) 计算两个要素或顶点 之间的相关性,如下 所示:
,由 Pearson 相关系数确定。基因调控网络表现出无标度拓扑结构36,其特征在于存在具有关键功能作用的枢纽基因。我们使用拓扑重叠度量 (TOM) 计算两个要素或顶点 之间的相关性,如下 所示:
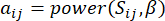 (1)
(1)
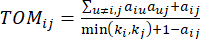 (2)
(2)
软阈值β是使用 WGCNA 软件包中的“pickSoft Threshold”函数计算的。应用幂指数函数 a ij,其中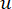 表示不包括 i 和 j 的基因,并表示
表示不包括 i 和 j 的基因,并表示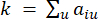 顶点连通性。WGCNA 使用常用的差异度量将转录组学特征的表达谱聚类到多个模块中 (
顶点连通性。WGCNA 使用常用的差异度量将转录组学特征的表达谱聚类到多个模块中 (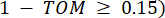 37.
37.
HealthModel 框架最初被设计为多任务学习架构26.该协议仅利用模型预训练任务来构建转录组mqTrans视图。用户可以选择使用额外的特定任务转录组样本在多任务图注意力网络下进一步完善预训练的 HealthModel。
特征选择和分类的技术细节
特征选择池实现了 11 种特征选择 (FS) 算法。其中,基于滤波器的FS算法有三种:使用最大信息系数(SK_mic)选择K个最佳特征,根据MIC的FPR(SK_fpr)选择K个特征,选择MIC错误发现率最高的K个特征(SK_fdr)。此外,三种基于树的 FS 算法使用具有 Gini 指数 (DT_gini)、自适应提升决策树 (AdaBoost) 和随机森林 (RF_fs) 的决策树来评估单个特征。该池还包含两种包装方法:使用线性支持向量分类器 (RFE_SVC) 的递归特征消除和使用逻辑回归分类器 (RFE_LR) 的递归特征消除。最后,包括两种嵌入算法:具有排名靠前的 L1 特征重要性值 (lSVC_L1) 的线性 SVC 分类器和具有排名靠前的 L1 特征重要性值 (LR_L1) 的逻辑回归分类器。
分类器池使用七种不同的分类器来构建分类模型。这些分类器包括线性支持向量机 (SVC)、高斯朴素贝叶斯 (GNB)、逻辑回归分类器 (LR)、k 最近邻(k 默认设置为 5 (KNN)、XGBoost、随机森林 (RF) 和决策树 (DT)。
可以在命令行中设置数据集的随机拆分为训练:测试子集。演示的示例使用火车的比率:test = 8:2。
