Das Transkriptom besteht aus der Expression aller Gene in einer Probe und kann mit Hochdurchsatztechnologien wie Microarray und RNA-seq1 profiliert werden. Die Expressionsniveaus eines Gens in einem Datensatz werden als transkriptomisches Merkmal bezeichnet, und die differentielle Repräsentation eines transkriptomischen Merkmals zwischen dem Phänotyp und der Kontrollgruppe definiert dieses Gen als Biomarker für diesen Phänotyp 2,3. Transkriptomische Biomarker wurden in großem Umfang bei der Untersuchung der Krankheitsdiagnose4, des biologischen Mechanismus5 und der Überlebensanalyse 6,7 usw. eingesetzt.
Genaktivitätsmuster in den gesunden Geweben enthalten wichtige Informationen über das Leben 8,9. Diese Muster bieten unschätzbare Erkenntnisse und dienen als ideale Referenzen für das Verständnis der komplexen Entwicklungsverläufe von gutartigen Erkrankungen10,11 und tödlichen Erkrankungen12. Gene interagieren miteinander, und Transkriptome stellen die endgültigen Expressionsniveaus nach ihren komplizierten Interaktionen dar. Solche Muster werden als transkriptionelles Regulationsnetzwerk13 und Stoffwechselnetzwerk14 usw. formuliert. Die Expression von Boten-RNAs (mRNAs) kann durch Transkriptionsfaktoren (TFs) und lange intergene nicht-kodierende RNAs (lincRNAs) transkriptionell reguliert werden15,16,17. Die konventionelle differentielle Expressionsanalyse ignorierte solche komplexen Geninteraktionen mit der Annahme der Unabhängigkeit zwischen den Merkmalen18,19.
Jüngste Fortschritte bei neuronalen Graphennetzen (GNNs) zeigen ein außerordentliches Potenzial bei der Extraktion wichtiger Informationen aus OMIC-basierten Daten für Krebsstudien20, z. B. die Identifizierung von Co-Expressionsmodulen21. Die angeborene Kapazität von GNNs macht sie ideal für die Modellierung der komplizierten Beziehungen und Abhängigkeiten zwischen Genen22,23.
Biomedizinische Studien konzentrieren sich oft auf die genaue Vorhersage eines Phänotyps im Vergleich zur Kontrollgruppe. Solche Aufgaben werden üblicherweise als binäre Klassifikationen24, 25, 26 formuliert. Hier werden die beiden Klassenbezeichnungen in der Regel als 1 und 0, wahr und falsch oder sogar positiv und negativ27 codiert.
Ziel dieser Studie war es, ein einfach zu verwendendes Protokoll zur Generierung der Transkriptionsregulation (mqTrans) eines Transkriptom-Datensatzes auf der Grundlage des vortrainierten Graph-Attention-Netzwerks (GAT) Referenzmodells bereitzustellen. Das Multitasking-GAT-Framework aus einer zuvor veröffentlichten Arbeit26 wurde verwendet, um transkriptomische Merkmale in die mqTrans-Merkmale umzuwandeln. Ein großer Datensatz gesunder Transkriptome der Xena-Plattform28 der University of California, Santa Cruz (UCSC) wurde verwendet, um das Referenzmodell (HealthModel) vorzutrainieren, das die Transkriptionsregulation von den regulatorischen Faktoren (TFs und lincRNAs) zu den Ziel-mRNAs quantitativ maß. Die generierte mqTrans-Ansicht könnte verwendet werden, um Vorhersagemodelle zu erstellen und dunkle Biomarker zu erkennen. Dieses Protokoll verwendet den Patientendatensatz für Dickdarmadenokarzinome (COAD) aus der Datenbank 29 des Cancer Genome Atlas (TCGA)29 als anschauliches Beispiel. In diesem Zusammenhang werden Patienten in den Stadien I oder II als negative Proben eingestuft, während Patienten in den Stadien III oder IV als positive Proben gelten. Die Verteilungen von dunklen und traditionellen Biomarkern über die 26 TCGA-Krebsarten hinweg werden ebenfalls verglichen.
Beschreibung der HealthModel-Pipeline
Die in diesem Protokoll verwendete Methodik basiert auf dem zuvor veröffentlichten Framework26, wie in Abbildung 1 skizziert. Zu Beginn müssen die Benutzer den Eingabedatensatz vorbereiten, ihn in die vorgeschlagene HealthModel-Pipeline einspeisen und mqTrans-Funktionen abrufen. Detaillierte Anweisungen zur Datenaufbereitung finden Sie in Abschnitt 2 des Protokollabschnitts. Anschließend haben Benutzer die Möglichkeit, mqTrans-Merkmale mit den ursprünglichen transkriptomischen Merkmalen zu kombinieren oder nur mit den generierten mqTrans-Merkmalen fortzufahren. Der erzeugte Datensatz wird dann einem Merkmalsauswahlprozess unterzogen, wobei die Benutzer die Flexibilität haben, ihren bevorzugten Wert für k in der k-fachen Kreuzvalidierung für die Klassifizierung zu wählen. Die primäre Bewertungsmetrik, die in diesem Protokoll verwendet wird, ist die Genauigkeit.
HealthModel26 kategorisiert die transkriptomischen Merkmale in drei verschiedene Gruppen: TF (Transkriptionsfaktor), lincRNA (lange intergene nicht-kodierende RNA) und mRNA (Boten-RNA). Die TF-Merkmale werden auf der Grundlage der im Human Protein Atlas30,31 verfügbaren Annotationen definiert. In dieser Arbeit werden die Annotationen von lincRNAs aus dem GTEx-Datensatz32 verwendet. Gene, die zu den Signalwegen der dritten Ebene in der KEGG-Datenbank33 gehören, werden als mRNA-Merkmale betrachtet. Es ist erwähnenswert, dass, wenn ein mRNA-Merkmal regulatorische Rollen für ein Zielgen aufweist, wie es in der TRRUST-Datenbank34 dokumentiert ist, es in die TF-Klasse umklassifiziert wird.
Dieses Protokoll generiert auch manuell die beiden Beispieldateien für die Gen-IDs der regulatorischen Faktoren (regulatory_geneIDs.csv) und der Ziel-mRNA (target_geneIDs.csv). Die paarweise Distanzmatrix zwischen den regulatorischen Merkmalen (TFs und lincRNAs) wird mit den Pearson-Korrelationskoeffizienten berechnet und mit der beliebten toolgewichteten Gen-Co-Expressions-Netzwerkanalyse (WGCNA)36 (adjacent_matrix.csv) geclustert. Benutzer können die HealthModel-Pipeline zusammen mit diesen Beispielkonfigurationsdateien direkt verwenden, um die mqTrans-Ansicht eines transkriptomischen Datensatzes zu generieren.
Technische Details von HealthModel
HealthModel stellt die komplizierten Beziehungen zwischen TFs und lincRNAs als Graph dar, wobei die Eingabemerkmale als die mit V bezeichneten Eckpunkte und eine als E bezeichnete Zwischenscheitelpunktkantenmatrix dienen. Jede Probe ist durch K-Regulationsmerkmale gekennzeichnet, die als VK×1 symbolisiert sind. Konkret umfasste der Datensatz 425 TFs und 375 lincRNAs, was zu einer Probendimensionalität von K = 425 + 375 = 800 führte. Um die Kantenmatrix E zu ermitteln, wurde in dieser Arbeit das beliebte Werkzeug WGCNA35 verwendet. Die paarweise Gewichtung, die zwei Scheitelpunkte verbindet, die als und 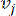 dargestellt werden
dargestellt werden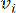 , wird durch den Pearson-Korrelationskoeffizienten bestimmt. Das genregulatorische Netzwerk weist eine skalenfreie Topologie36 auf, die durch das Vorhandensein von Hub-Genen mit zentralen funktionellen Rollen gekennzeichnet ist. Wir berechnen die Korrelation zwischen zwei Features oder Stützpunkten und , indem wir das topologische Überlappungsmaß (TOM) wie folgt verwenden:
, wird durch den Pearson-Korrelationskoeffizienten bestimmt. Das genregulatorische Netzwerk weist eine skalenfreie Topologie36 auf, die durch das Vorhandensein von Hub-Genen mit zentralen funktionellen Rollen gekennzeichnet ist. Wir berechnen die Korrelation zwischen zwei Features oder Stützpunkten und , indem wir das topologische Überlappungsmaß (TOM) wie folgt verwenden:
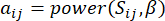 (1)
(1)
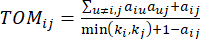 (2)
(2)
Der weiche Schwellenwert β wird mit der Funktion “pickSoft Threshold” aus dem WGCNA-Paket berechnet. Es wird die Potenzexponentialfunktion aij angewendet, wobei 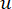 ein Gen ohne i und j steht und
ein Gen ohne i und j steht und 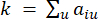 die Vertex-Konnektivität darstellt. WGCNA gruppiert die Expressionsprofile der transkriptomischen Merkmale in mehrere Module unter Verwendung eines häufig verwendeten Unähnlichkeitsmaßes (
die Vertex-Konnektivität darstellt. WGCNA gruppiert die Expressionsprofile der transkriptomischen Merkmale in mehrere Module unter Verwendung eines häufig verwendeten Unähnlichkeitsmaßes (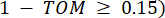 37.
37.
Das HealthModel-Framework wurde ursprünglich als Multitasking-Lernarchitekturkonzipiert 26. Dieses Protokoll verwendet nur die Modell-Pre-Training-Aufgabe für die Konstruktion der transkriptomischen mqTrans-Ansicht. Der Benutzer kann sich dafür entscheiden, das vortrainierte HealthModel im Rahmen des Multitasking-Graph-Aufmerksamkeitsnetzwerks mit zusätzlichen aufgabenspezifischen transkriptomischen Proben weiter zu verfeinern.
Technische Details der Merkmalsauswahl und -klassifizierung
Der Feature-Selection-Pool implementiert elf Feature-Selection-Algorithmen (FS). Darunter sind drei filterbasierte FS-Algorithmen: die Auswahl der besten K-Merkmale unter Verwendung des maximalen Informationskoeffizienten (SK_mic), die Auswahl von K-Merkmalen basierend auf der FPR von MIC (SK_fpr) und die Auswahl von K-Merkmalen mit der höchsten False-Discovery-Rate von MIC (SK_fdr). Darüber hinaus bewerten drei baumbasierte FS-Algorithmen einzelne Merkmale mithilfe eines Entscheidungsbaums mit dem Gini-Index (DT_gini), adaptiven verstärkten Entscheidungsbäumen (AdaBoost) und Random Forest (RF_fs). Der Pool enthält auch zwei Wrappermethoden: die rekursive Featureeliminierung mit dem linearen Unterstützungsvektorklassifikator (RFE_SVC) und die rekursive Featureeliminierung mit dem logistischen Regressionsklassifikator (RFE_LR). Schließlich sind zwei Einbettungsalgorithmen enthalten: der lineare SVC-Klassifikator mit den am höchsten bewerteten L1-Merkmalsbedeutungswerten (lSVC_L1) und der logistische Regressionsklassifikator mit den am höchsten bewerteten L1-Merkmalswichtigkeitswerten (LR_L1).
Der Klassifikatorpool verwendet sieben verschiedene Klassifikatoren, um Klassifizierungsmodelle zu erstellen. Zu diesen Klassifikatoren gehören Linear Support Vector Machine (SVC), Gaußian Naïve Bayes (GNB), Logistic Regression Classifier (LR), k-Nearest Neighbor, wobei k standardmäßig auf 5 gesetzt ist (KNN), XGBoost, Random Forest (RF) und Decision Tree (DT).
Die zufällige Aufteilung des Datensatzes in die train: Test-Teilmengen kann in der Befehlszeile festgelegt werden. Im gezeigten Beispiel wird das Verhältnis von train: test = 8:2 verwendet.
