שעתוק מורכב מהביטויים של כל הגנים בדגימה ועשוי להיות פרופיל על ידי טכנולוגיות תפוקה גבוהה כמו microarray ו- RNA-seq1. רמות הביטוי של גן אחד במערך נתונים נקראות תכונת שעתוק (transcriptomic feature), והייצוג הדיפרנציאלי של תכונת שעתוק בין קבוצת הפנוטיפ וקבוצת הביקורת מגדיר גן זה כסמן ביולוגי של פנוטיפזה 2,3. סמנים ביולוגיים שעתוק שימשו באופן נרחב בחקירות של אבחון מחלות4, מנגנון ביולוגי5, וניתוח הישרדות 6,7 וכו ‘.
דפוסי הפעילות הגנטית ברקמות הבריאות נושאים מידע חיוני על החיים 8,9. דפוסים אלה מציעים תובנות שלא יסולא בפז ומשמשים כמקורות אידיאליים להבנת מסלולי ההתפתחות המורכבים של הפרעות שפירות10,11 ומחלות קטלניות12. גנים מתקשרים זה עם זה, ותעתיקים מייצגים את רמות הביטוי הסופיות לאחר האינטראקציות המסובכות שלהם. דפוסים אלה מנוסחים כרשת ויסות שעתוק13 ורשת מטבוליזם14 וכו ‘. הביטויים של רנ”א שליח (mRNAs) ניתנים לשעתוק על ידי גורמי שעתוק (TFs) ורנ”א אינטרגני ארוך שאינו מקודד (lincRNAs)15,16,17. ניתוח ביטוי דיפרנציאלי קונבנציונלי התעלם מאינטראקציות גנים מורכבות כאלה עם ההנחה של עצמאות בין תכונות18,19.
ההתקדמות האחרונה ברשתות עצביות גרפיות (GNNs) מדגימה פוטנציאל יוצא דופן בחילוץ מידע חשוב מנתונים מבוססי OMIC עבור מחקרי סרטן20, למשל, זיהוי מודולי ביטוי משותף21. היכולת המולדת של GNNs הופכת אותם לאידיאליים למידול היחסים המורכבים והתלות בין גנים22,23.
מחקרים ביו-רפואיים מתמקדים לעתים קרובות בניבוי מדויק של פנוטיפ כנגד קבוצת הביקורת. משימות כאלה מנוסחות בדרך כלל כסיווגים בינאריים 24,25,26. כאן, שתי תוויות המחלקה מקודדות בדרך כלל כ- 1 ו- 0, נכון ולא נכון, או אפילו חיובי ושלילי27.
מחקר זה נועד לספק פרוטוקול קל לשימוש ליצירת תצוגת ויסות התמלול (mqTrans) של ערכת נתוני תמלול המבוססת על מודל הייחוס של רשת תשומת הלב הגרפים (GAT) שהוכשר מראש. מסגרת GAT מרובת המשימות מעבודה26 שפורסמה בעבר שימשה להמרת תכונות תעתיק לתכונות mqTrans. מערך נתונים גדול של תעתיקים בריאים מאוניברסיטת קליפורניה, סנטה קרוז (UCSC) Xenaפלטפורמה 28 שימש לאימון מראש של מודל הייחוס (HealthModel), אשר מדד כמותית את תקנות השעתוק מהגורמים הרגולטוריים (TFs ו- lincRNAs) ל- mRNA המטרה. תצוגת mqTrans שנוצרה יכולה לשמש לבניית מודלים לחיזוי ולזיהוי סמנים ביולוגיים כהים. פרוטוקול זה משתמש במערך הנתונים של חולי אדנוקרצינומה של המעי הגס (COAD) מתוך מסד הנתונים29 של אטלס גנום הסרטן (TCGA) כדוגמה להמחשה. בהקשר זה, חולים בשלבים I או II מסווגים כדגימות שליליות, ואילו אלה בשלבים III או IV נחשבים לדגימות חיוביות. כמו כן מושווים ההתפלגויות של סמנים ביולוגיים כהים ומסורתיים על פני 26 סוגי סרטן TCGA.
תיאור הצינור של HealthModel
המתודולוגיה המיושמת בפרוטוקול זה מבוססת על מסגרת26 שפורסמה בעבר, כמתואר באיור 1. כדי להתחיל, המשתמשים נדרשים להכין את ערכת נתוני הקלט, להזין אותו לתוך צינור HealthModel המוצע, ולקבל תכונות mqTrans. הוראות מפורטות להכנת נתונים מפורטות מפורטות בסעיף 2 בסעיף הפרוטוקול. לאחר מכן, למשתמשים יש אפשרות לשלב תכונות mqTrans עם תכונות התמליל המקוריות או להמשיך עם תכונות mqTrans שנוצרו בלבד. לאחר מכן, ערכת הנתונים המופקת כפופה לתהליך בחירת תכונות, כאשר למשתמשים יש את הגמישות לבחור את הערך המועדף עליהם עבור k באימות צולב k-fold לסיווג. מדד ההערכה העיקרי המשמש בפרוטוקול זה הוא דיוק.
HealthModel26 מסווג את תכונות השעתוק לשלוש קבוצות נפרדות: TF (גורם שעתוק), lincRNA (RNA אינטרגני ארוך שאינו מקודד) ו- mRNA (רנ”א שליח). תכונות TF מוגדרות בהתבסס על הביאורים הזמינים באטלס החלבונים האנושי30,31. עבודה זו משתמשת בביאורים של lincRNAs מתוך מערך הנתונים GTEx32. גנים השייכים למסלולים ברמה השלישית במסד הנתוניםKEGG 33 נחשבים לתכונות mRNA. ראוי לציין כי אם תכונת mRNA מציגה תפקידי בקרה עבור גן מטרה כפי שמתועד במסד הנתונים TRRUST34, הוא מסווג מחדש למחלקת TF.
פרוטוקול זה גם מייצר באופן ידני את שני קבצי הדוגמה עבור מזהי הגנים של גורמים רגולטוריים (regulatory_geneIDs.csv) ו- mRNA מטרה (target_geneIDs.csv). מטריצת המרחק הזוגי בין תכונות הבקרה (TFs ו- lincRNAs) מחושבת על ידי מקדמי המתאם של פירסון ומקובצת על ידי הכלי הפופולרי ניתוח רשת ביטוי משותף של גנים (WGCNA)36 (adjacent_matrix.csv). משתמשים יכולים להשתמש ישירות בצינור HealthModel יחד עם קבצי תצורה לדוגמה אלה כדי ליצור את תצוגת mqTrans של ערכת נתונים תמלולית.
פרטים טכניים של HealthModel
HealthModel מייצג את היחסים המורכבים בין TFs ו- lincRNA כגרף, כאשר תכונות הקלט משמשות כקודקודים המסומנים ב- V ומטריצת קצה בין קודקודים המסומנת כ- E. כל מדגם מאופיין בתכונות רגולטוריות K , המסומלות כ – VK×1. באופן ספציפי, מערך הנתונים הקיף 425 TFs ו 375 lincRNAs, וכתוצאה מכך ממדיות מדגם של K = 425 + 375 = 800. כדי להקים את מטריצת הקצה E, עבודה זו השתמשה בכלי הפופולרי WGCNA35. המשקל הזוגי המקשר בין שני קודקודים המיוצגים כ 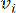 – ו
– ו 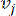 – , נקבע על ידי מקדם המתאם של פירסון. רשת הבקרה הגנטית מציגה טופולוגיה36 נטולת קנה מידה, המאופיינת בנוכחות גנים רכזתיים בעלי תפקידים פונקציונליים מרכזיים. אנו מחשבים את המתאם בין שתי תכונות או קודקודים, ו – , באמצעות מדד החפיפה הטופולוגית (TOM) באופן הבא:
– , נקבע על ידי מקדם המתאם של פירסון. רשת הבקרה הגנטית מציגה טופולוגיה36 נטולת קנה מידה, המאופיינת בנוכחות גנים רכזתיים בעלי תפקידים פונקציונליים מרכזיים. אנו מחשבים את המתאם בין שתי תכונות או קודקודים, ו – , באמצעות מדד החפיפה הטופולוגית (TOM) באופן הבא:
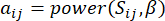 (1)
(1)
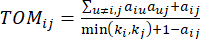 (2)
(2)
β הסף הרך מחושב באמצעות הפונקציה ‘pickSoft Threshold’ מחבילת WGCNA. הפונקציה מעריכית החזקה aij מוחלת, כאשר 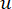 מייצג גן שאינו כולל i ו– j, ומייצג
מייצג גן שאינו כולל i ו– j, ומייצג 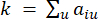 את קישוריות הקודקוד. WGCNA מקבץ את פרופילי הביטוי של תכונות התעתיק למודולים מרובים באמצעות מדד אי-דמיון נפוץ (
את קישוריות הקודקוד. WGCNA מקבץ את פרופילי הביטוי של תכונות התעתיק למודולים מרובים באמצעות מדד אי-דמיון נפוץ (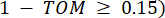 37.
37.
מסגרת HealthModel תוכננה במקור כארכיטקטורת למידה מרובת משימות26. פרוטוקול זה משתמש רק במשימת האימון מראש של המודל לבניית תצוגת mqTrans התעתוק. המשתמש יכול לבחור לחדד עוד יותר את HealthModel שהוכשר מראש תחת רשת הקשב של גרף ריבוי המשימות עם דגימות תמלול ספציפיות למשימה נוספות.
פרטים טכניים של בחירת תכונות וסיווגן
מאגר בחירת התכונות מיישם אחד-עשר אלגוריתמים לבחירת תכונות (FS). ביניהם, שלושה הם אלגוריתמי FS מבוססי מסנן: בחירת התכונות הטובות ביותר של K באמצעות מקדם המידע המרבי (SK_mic), בחירת תכונות K בהתבסס על FPR של מיקרופון (SK_fpr), ובחירת תכונות K עם שיעור גילוי השווא הגבוה ביותר של מיקרופון (SK_fdr). בנוסף, שלושה אלגוריתמי FS מבוססי עצים מעריכים תכונות בודדות באמצעות עץ החלטה עם מדד ג’יני (DT_gini), עצי החלטה מוגברת אדפטיבית (AdaBoost) ויער אקראי (RF_fs). המאגר משלב גם שתי שיטות עטיפה: חיסול תכונות רקורסיבי עם מסווג וקטור תמיכה ליניארי (RFE_SVC) וביטול תכונות רקורסיבי עם מסווג רגרסיה לוגיסטית (RFE_LR). לבסוף, שני אלגוריתמי הטבעה כלולים: מסווג SVC ליניארי עם ערכי חשיבות התכונות L1 המדורגים במקום הראשון (lSVC_L1) ומסווג רגרסיה לוגיסטית עם ערכי חשיבות התכונות L1 המדורגים במקום הראשון (LR_L1).
מאגר המסווגים מעסיק שבעה מסווגים שונים לבניית מודלים לסיווג. מסווגים אלה כוללים מכונת וקטור תמיכה ליניארית (SVC), גאוסיאן נאיבי בייס (GNB), מסווג רגרסיה לוגיסטית (LR), k השכן הקרוב ביותר, כאשר k מוגדר ל- 5 כברירת מחדל (KNN), XGBoost, יער אקראי (RF) ועץ החלטה (DT).
הפיצול האקראי של מערך הנתונים לרכבת: ניתן להגדיר תת-קבוצות בדיקה בשורת הפקודה. הדוגמה המודגמת משתמשת ביחס של רכבת: מבחן = 8: 2.
