Транскриптом состоит из экспрессии всех генов в образце и может быть профилирован с помощью высокопроизводительных технологий, таких как микрочип и РНК-секвенирование1. Уровни экспрессии одного гена в наборе данных называются транскриптомным признаком, а дифференциальное представление транскриптомного признака между фенотипом и контрольной группами определяет этот ген как биомаркер этого фенотипа 2,3. Транскриптомные биомаркеры широко используются в исследованиях диагностики заболеваний4, биологического механизма5, анализа выживаемости 6,7 и т.д.
Паттерны активности генов в здоровых тканях несут важнейшую информацию о жизни 8,9. Эти закономерности дают неоценимую информацию и служат идеальными справочными материалами для понимания сложных траекторий развития доброкачественных заболеваний10,11 и смертельных заболеваний12. Гены взаимодействуют друг с другом, и транскриптомы представляют собой конечные уровни экспрессии после их сложных взаимодействий. Такие паттерны формулируются как транскрипционная регуляционная сеть13 и метаболическая сеть14 и др. Экспрессия матричных РНК (мРНК) может транскрипционно регулироваться транскрипционными факторами (ТФ) и длинными межгенными некодирующими РНК (линкРНК)15,16,17. Традиционный анализ дифференциальной экспрессии игнорировал такие сложные взаимодействия генов с предположением о независимости между признаками18,19.
Недавние достижения в области графовых нейронных сетей (GNN) демонстрируют необычайный потенциал в извлечении важной информации из данных, основанных на OMIC, для исследованийрака20, например, идентификация модулей коэкспрессии21. Врожденная способность GNN делает их идеальными для моделирования сложных взаимоотношений и зависимостей между генами22,23.
Биомедицинские исследования часто сосредоточены на точном прогнозировании фенотипа по сравнению с контрольной группой. Такие задачи обычно формулируются в виде бинарных классификаций 24,25,26. Здесь две метки классов обычно кодируются как 1 и 0, true и false или даже positive и negative27.
Это исследование было направлено на предоставление простого в использовании протокола для создания представления транскрипционной регуляции (mqTrans) набора данных транскриптома на основе предварительно обученной эталонной модели сети графового внимания (GAT). Для преобразования транскриптомных признаков в признаки mqTrans был использован многозадачный фреймворк GAT из ранее опубликованной работы26 . Большой набор данных здоровых транскриптомов из платформы Xena28 Калифорнийского университета в Санта-Крузе (UCSC) был использован для предварительного обучения референсной модели (HealthModel), которая количественно измеряла регуляции транскрипции от регуляторных факторов (ТФ и линкРНК) до целевых мРНК. Сгенерированное представление mqTrans может быть использовано для построения моделей прогнозирования и обнаружения темных биомаркеров. В этом протоколе в качестве иллюстративного примера используется набор данных пациентов с аденокарциномой толстой кишки (COAD) из базы данных29 Атласа генома рака (TCGA). В этом контексте пациенты на I или II стадиях классифицируются как отрицательные образцы, в то время как пациенты на III или IV стадиях считаются положительными образцами. Также сравнивается распределение темновых и традиционных биомаркеров по 26 типам рака TCGA.
Описание конвейера HealthModel
Методология, используемая в этом протоколе, основана на ранее опубликованной структуре26, как показано на рисунке 1. Для начала пользователям необходимо подготовить входной набор данных, передать его в предлагаемый конвейер HealthModel и получить функции mqTrans. Подробные инструкции по подготовке данных приведены в разделе 2 раздела протокола. После этого у пользователей есть возможность комбинировать признаки mqTrans с исходными транскриптомными признаками или продолжать только с сгенерированными признаками mqTrans. Затем полученный набор данных подвергается процессу выбора признаков, при этом пользователи могут выбрать предпочтительное значение для k в k-кратной перекрестной проверке для классификации. Основным оценочным показателем, используемым в этом протоколе, является точность.
HealthModel26 классифицирует транскриптомные признаки по трем отдельным группам: TF (транскрипционный фактор), lincRNA (длинная межгенная некодирующая РНК) и mRNA (матричная РНК). Признаки TF определяются на основе аннотаций, доступных в Атласе белков человека30,31. В данной работе используются аннотации линкРНК из набора данных GTEx32. Гены, принадлежащие к путям третьего уровня в базе данных KEGG33, рассматриваются как признаки мРНК. Стоит отметить, что если признак мРНК проявляет регуляторную роль для гена-мишени, как это задокументировано в базе данных TRRUST34, он реклассифицируется в класс TF.
Этот протокол также вручную генерирует два файла примеров для идентификаторов генов регуляторных факторов (regulatory_geneIDs.csv) и мРНК-мишеней (target_geneIDs.csv). Матрица попарных расстояний между регуляторными признаками (ТФ и линкРНК) вычисляется с помощью коэффициентов корреляции Пирсона и кластеризуется с помощью популярного инструментального взвешенного сетевого анализа генной коэкспрессии (WGCNA)36 (adjacent_matrix.csv). Пользователи могут напрямую использовать конвейер HealthModel вместе с этими примерами файлов конфигурации для создания представления mqTrans набора транскриптомных данных.
Технические характеристики HealthModel
HealthModel представляет сложные взаимосвязи между ТФ и линкРНК в виде графа, где входные объекты служат вершинами, обозначенными V , и матрицей межвершинных ребер, обозначенной как E. Каждый образец характеризуется К-регуляторными признаками, обозначаемыми как VK×1. В частности, набор данных включал 425 ТФ и 375 линкРНК, в результате чего размерность выборки составила K = 425 + 375 = 800. Для установления матрицы кромок E в этой работе использовался популярный инструмент WGCNA35. Попарный вес, связывающий две вершины, представленные как 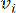 и
и 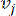 , определяется коэффициентом корреляции Пирсона. Генная регуляторная сеть имеет безмасштабную топологию36, характеризующуюся присутствием генов-концентраторов с ключевыми функциональными ролями. Мы вычисляем корреляцию между двумя объектами или вершинами, и , используя меру топологического перекрытия (TOM) следующим образом:
, определяется коэффициентом корреляции Пирсона. Генная регуляторная сеть имеет безмасштабную топологию36, характеризующуюся присутствием генов-концентраторов с ключевыми функциональными ролями. Мы вычисляем корреляцию между двумя объектами или вершинами, и , используя меру топологического перекрытия (TOM) следующим образом:
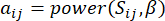 (1)
(1)
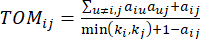 (2)
(2)
Мягкий пороговый β вычисляется с помощью функции ‘pickSoft Threshold’ из пакета WGCNA. Применяется степенная экспоненциальная функция a ij, где 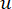 представляет ген, исключая i и j, и
представляет ген, исключая i и j, и 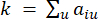 представляет связность вершин. WGCNA кластеризует профили экспрессии транскриптомных признаков в несколько модулей, используя широко используемую меру несходства (
представляет связность вершин. WGCNA кластеризует профили экспрессии транскриптомных признаков в несколько модулей, используя широко используемую меру несходства (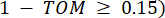 37.
37.
Фреймворк HealthModel изначально разрабатывался как многозадачная архитектура обучения26. Этот протокол использует только задачу предварительного обучения модели для построения транскриптомного представления mqTrans. Пользователь может дополнительно усовершенствовать предварительно обученную модель HealthModel в многозадачной графовой сети внимания с помощью дополнительных транскриптомных образцов, специфичных для конкретной задачи.
Технические сведения о выборе и классификации функций
Пул выбора признаков реализует одиннадцать алгоритмов выбора признаков (FS). Среди них три алгоритма ФС на основе фильтров: выбор K лучших признаков с использованием максимального коэффициента информации (SK_mic), выбор K признаков на основе FPR MIC (SK_fpr) и выбор K объектов с наибольшим уровнем ложного обнаружения MIC (SK_fdr). Кроме того, три древовидных алгоритма ФС оценивают отдельные признаки с помощью дерева решений с индексом Джини (DT_gini), адаптивного дерева решений (AdaBoost) и случайного леса (RF_fs). Пул также включает в себя два метода-оболочки: рекурсивное исключение признаков с помощью классификатора линейных опорных векторов (RFE_SVC) и рекурсивное исключение признаков с классификатором логистической регрессии (RFE_LR). Наконец, включены два алгоритма внедрения: линейный классификатор SVC с самыми ранжированными значениями важности признаков L1 (lSVC_L1) и классификатор логистической регрессии с самыми ранжированными значениями важности признаков L1 (LR_L1).
Пул классификаторов использует семь различных классификаторов для построения моделей классификации. Эти классификаторы включают в себя линейный метод опорных векторов (SVC), наивный байесовский метод Гаусса (GNB), классификатор логистической регрессии (LR), k-ближайшего соседа, с k, установленным в 5 по умолчанию (KNN), XGBoost, случайный лес (RF) и дерево решений (DT).
Случайное разбиение датасета на обучающие: тестовые подмножества можно задать в командной строке. В приведенном примере используется соотношение train: test = 8:2.
