Determination of Intracellular Ascorbate in Cultured Suspension Cells
In the first assay (Figure 1), intracellular ascorbate is determined, following ascorbate-specific (i.e. AO-sensitive) reduction of ferricyanide to ferrocyanide, using the highly sensitive determination of ferrocyanide by a previously published procedure37. The detection of ascorbate is based on the colorimetric chelation of ferrous iron that is generated by the ascorbate-dependent reduction of ferricyanide to ferrocyanide, followed by the reduction of ferric to ferrous iron by ferrocyanide. As supraphysiological concentrations of some divalent metal ions (e.g. Cu2+, Co2+ and Zn2+) can interfere, presumably by competing with ferrous iron for chelation by ferene-S, appropriate precautions should be taken under such conditions37.
Figure 2 shows a typical standard curve for a set of ascorbate standards 0-20 μM (or 0-2 nmole ascorbate per well of the 96-well plate; see step 6.5 in the "Protocol".). Although not shown in this figure, linearity is maintained up to 8 nmole ascorbate per well (i.e. a net A593 nm of ~1.6), which corresponds to a sample ascorbate concentration of ~80 μM. The assay can be used to successfully detect ascorbate levels below 0.25 nmole ascorbate per well (2.5 μM sample ascorbate concentration).
K562 cells rapidly accumulate intracellular ascorbate from extracellular DHA, but not ascorbate (see Figure 3; reproduced with permission from Lane and Lawen 200819). In this representative experiment, PBS-washed K562 cells (4 × 106 cells/ml) were incubated in PBS with 500 μM of either ascorbate (Asc), DHA, DHA + 50 μM cytochalasin B (DHA+CB), Asc + 5 mM ferricyanide (Asc/FIC) or Asc + 50 U/ml AO (Asc/AO) for 30 min at 37 °C. Intracellular ascorbate was determined as described in the "Protocol".
DHA uptake by K562 cells occurs by facilitative glucose transporter (GLUT)-mediated transport (see Figure 4; reproduced with permission from Lane and Lawen 200942). To assess the involvement of GLUTs in DHA uptake in this representative experiment, K562 cells were incubated with increasing concentrations of cytochalasin B (CB) or dihydrocytochalasin B (H2CB) dissolved in MBS containing 0.5% ethanol for 15 min at 37 °C prior to incubation with 400 μM DHA for 30 min at 37 °C in the same medium (Fig. 4A). Cells were then washed three times in 100 volumes of ice-cold MBS and their intracellular ascorbate determined as described in the “Protocol”. It is important to note while both CB and H2CB inhibit motile processes at micromolar concentrations (1-100 μM), only CB, which differs from H2CB by the presence of single double bond, inhibits GLUT-dependent transport at low micromolar concentrations (1-10 μM)43. Therefore, as DHA uptake was inhibitable by CB with an IC50 < 2.5 μM, but was not inhibitable by H2CB, DHA uptake probably occurs by GLUT-mediated transport38. Alternatively, in Figure 4B, washed cells were exposed to increasing concentrations of the GLUT-transportable, but non-metabolizable glucose analog 3-O-methyl-D-glucose (3-OMG) or the non-GLUT-transportable glucose stereoisomer L-glucose prior to incubation with DHA as in panel A. Again the results indicate GLUT involvement in DHA import as inhibition of intracellular ascorbate accumulation occurs only in the presence of the GLUT-transportable glucose analog.
Determination of Ascorbate-Efflux from Adherent Cells
In the second assay, the rate of ascorbate-efflux from cultured cells can be determined. This specific assay is important as the release of ascorbate from cells appears to be crucial for the ascorbate-regulated cellular uptake of non-transferrin-bound iron by cells19,20. The uptake of non-transferrin-bound iron is considered to be relevant to the pathophysiology of iron-overload disorders such as the hemochromatoses22, as well as to astrocyte-neuron iron exchange and homeostasis in the mammalian brain22. Indeed, we have recently shown that the major excitatory neurotransmitter, L-glutamate, triggers the release of ascorbate from astrocytes in a manner that depends on L-glutamate uptake by GLAST and subsequent cellular swelling that triggers the release of ascorbate by putative VSOACs32. In analogy, ascorbate release from ascorbate-loaded astrocytes can be stimulated by the excitatory amino acid, L-aspartate, but not the non-excitatory amino acid L-glutamine. This effect is depicted in Figure 5 (reproduced with permission from Lane and Lawen 201232), which shows dose-response curves for the stimulation of ascorbate (AA) release by aspartate (closed circles) and glutamine (open circles) from primary cultures of ascorbate-loaded mouse astrocytes.
It is important to discuss how this ascorbate-efflux assay differs from the assay provided for the determination of intracellular ascorbate. The major difference lies in the fact that the level of ascorbate remaining in solution is not determined by a chemical reaction conducted at the end of a given time point, as is the case for the intracellular ascorbate determination method. Instead, a "reductive signature" is accumulated during the period in which ascorbate is released from cells. This reductive signature is captured in the form of the ascorbate-dependent reduction of extracellular ferric citrate followed by the rapid chelation of the ferrous iron as a large extracellular and membrane-impermeant [Fe(II)(ferene-S)3]4- chelate. Ferene-S can be considered similarly membrane-impermeant over the short time course of the assay. The levels of this chromogenic complex can then be determined as an endpoint measurement. As only the AO-sensitive levels of this chelate are determined, the assay provides a high-degree of specificity for determination of extracellular L-ascorbate.
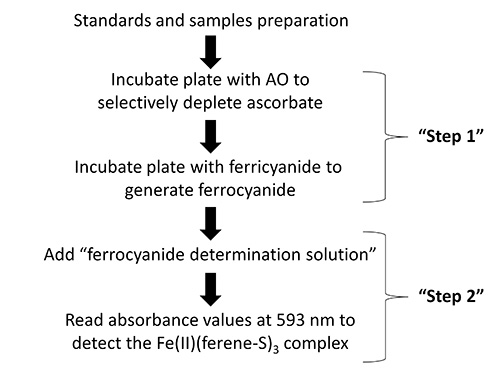
Figure 1. Flow diagram showing the key steps in the protocol for the determination of intracellular ascorbate.
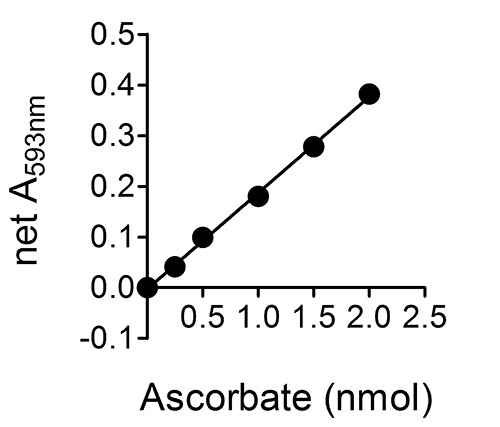
Figure 2. A typical standard curve for a set of ascorbate standards 0-20 μM (or 0-2 nmole ascorbate per well of the 96-well plate; see step 6.5 in the "Protocol".). Error bars are not shown as they are within the range occupied by the symbols.
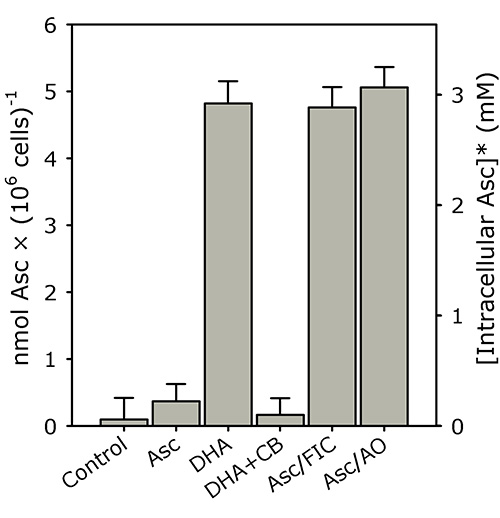
Figure 3. K562 cells rapidly accumulate intracellular ascorbate from extracellular DHA, but not ascorbate. PBS-washed K562 cells (4 × 106 cells/ml) were incubated in PBS with 500 μM of either ascorbate (Asc), DHA, DHA + 50 μM cytochalasin B (DHA+CB), Asc + 5 mM ferricyanide (Asc/FIC) or Asc + 50 U/ml AO (Asc/AO) for 30 min at 37 °C. Intracellular ascorbate was determined as described in the "Protocol". The results shown are means of three individual experiments (+ SD). *The intracellular ascorbate concentration was estimated using a pre-determined intracellular water space for K562 cells (i.e. ~1.6 μl/106 cells)19,20. This figure has been reproduced with permission from Lane and Lawen 200819.
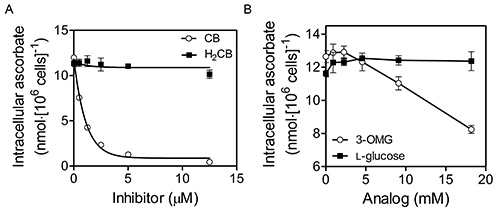
Figure 4. DHA uptake by K562 cells occurs by facilitative glucose transporter (GLUT)-mediated transport. K562 cells that had been grown to 6-8 × 106 cells/ml in RPMI + 10% (v/v) fetal bovine serum at 37 °C, 5% CO2 and 95% air were initially washed three times with MBS. Washed cells were then exposed to increasing concentrations of cytochalasin B (CB; Sigma) or dihydrocytochalasin B (H2CB) dissolved in Mops-buffered saline (MBS; 137 mM NaCl, 2.7 mM KCl, 15 mM MOPS-Na+, pH 7.3) containing 0.5% ethanol prior to incubation with 400 μM DHA for 30 min at 37 °C (A). Cells were then washed three times in 100 volumes of cold MBS and their intracellular ascorbate determined as described in the “Protocol”. As DHA uptake was inhibitable by CB, but not H2CB, DHA uptake occurs by GLUT-mediated transport. Alternatively, in (B), washed cells were exposed to increasing concentrations of the GLUT-transportable, but non-metabolizable glucose analog 3-O-methyl-D-glucose (3-OMG) or the non-GLUT-transportable glucose stereoisomer L-glucose during incubation with DHA as in (A). Again the results indicate GLUT involvement in DHA uptake. This figure has been reproduced with permission from Lane and Lawen 200842.
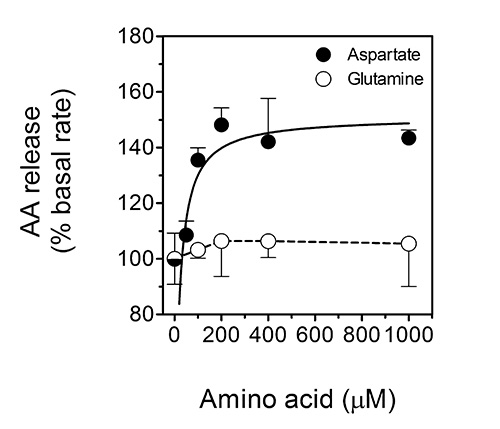
Figure 5. Ascorbate release from ascorbate-loaded astrocytes is stimulated by the excitatory amino acid L-aspartate, but not the non-excitatory amino acid L-glutamine. This figure shows dose-response curves for the stimulation of ascorbate (AA) release by aspartate (closed circles) and glutamine (open circles) from primary cultures of ascorbate-loaded mouse astrocytes. The data shown are means (± SD) of three experiments. P < 0.001 vs. the 'basal' condition. This figure has been reproduced with permission from Lane and Lawen 201232.