To elucidate the molecular mechanism of CP, it is necessary to identify the cellular compartments, where exogenous antigens undergo ERAD-like transport and processing. While observations by immunofluorescent microscopy or by electron microscopy identified the cellular compartment where exogenous antigens accumulated16,17,18,19,30,31,32,33, the cellular compartments for ERAD-like processing of exogenous antigens are not clearly defined. Recently it was shown that non-classical endosomes with ER resident molecules were responsible for CP34, but these cellular compartments were unpurified. The difficulty in isolating and purifying the cellular compartments can be attributed to the fact that exogenous antigens are localized both in the endosome and ER-like compartments and that there is no identifying molecule for these endocytic compartments other than exogenous antigens. However, the condition of exogenous antigens undergoing ERAD-like transport is different from the steady state; in the transport across lipid bimolecular membrane, exogenous antigens penetrate the membrane via translocons, such as Sec61 (Figure 2A). Thus, when using bOVA as an exogenous antigen, bOVA should be associated with Sec61. Since membrane-associated bOVA specifically bound with SA, the microsome prepared from DC2.4, which was pretreated with bOVA, could be isolated distinctively by SA-magnetic beads (Figure 2A). Then equivalent amounts of proteins from purified microsomes with or without pre-incubation of bOVA were resolved by SDS-PAGE followed by silver staining and Western blotting with SA-HRP (Figure 2B, 2C). As shown in Figure 2B and Figure 2C, the isolated microsomes contained several unique proteins that were purified dependent upon exogenously added bOVA and SA-magnetic beads. In addition to these unique proteins, isolated microsomes also contained nonspecific proteins, which bound to SA-magnetic beads with or without bOVA. Treatment of the microsome with trypsin before purification by the magnet prevented the purification of microsomes (Figure 2D), indicating that the purification methods depended on the presence of membrane-penetrating bOVA.
The purified microsomes showed the ability to ubiquitinate the incorporated bOVA in vitro, under the presence of RLs (Figure 3A). The amounts of bOVA and poly-ubiquitinated bOVA were augmented in the presence of MG132 (Figure 3B), indicating that the incorporated bOVA was processed by the ERAD system and that our purified microsomes contained ERAD machinery proteins.
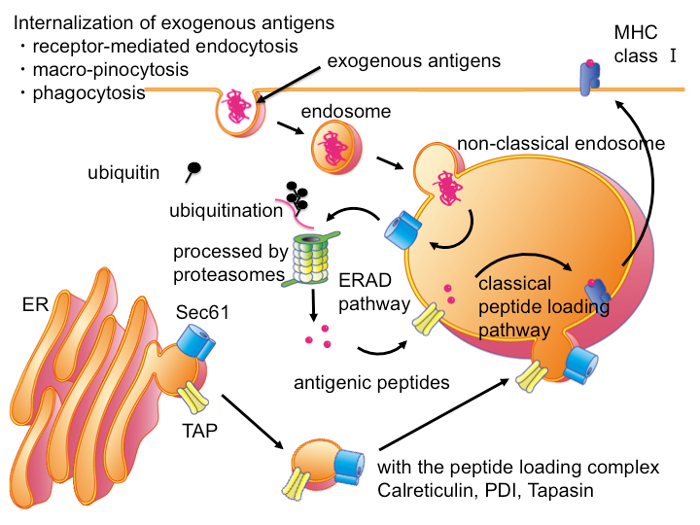
Figure 1: Intracellular Pathways for CP in DCs. In DCs, exogenous antigens are transported into non-classical endocytic compartments, which also contain ER-resident molecules in addition to molecules of the classical late endosome. In this compartment, exogenous antigens are exported into cytosol through translocons such as Sec61. In the cytosol, exogenous antigens are processed by the ubiquitin-proteasome system into antigenic peptides as ERAD substrates. Antigenic peptides are transported into same or adjacent non-classical endocytic compartments, or adjacent ER through TAP transporter and then loaded on the MHC I molecules by PLC. Please click here to view a larger version of this figure.
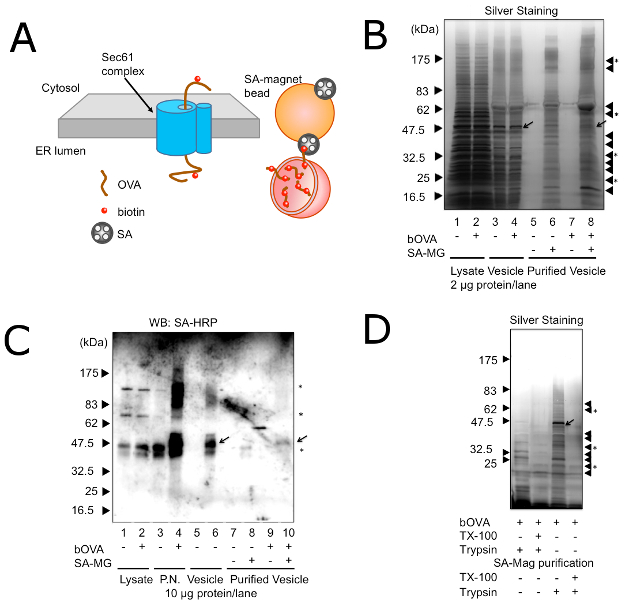
Figure 2: Purification of Microsomes with bOVA Undergoing ERAD. (A) A schematic model of purification of microsomes with bOVA undergoing ERAD. bOVA is associated with the membrane through the Sec61 translocon and targeted with SA-magnetic beads. (B) Microsomes with (+) or without (-) prior addition of bOVA were purified with (+) or without (-) SA-magnetic beads. Proteins (2 µg) or corresponding volumes of purified proteins were resolved on 7.5 – 15% SDS-PAGE, and silver staining was used to visualize protein bands. Triangles on the right side indicate nonspecific proteins binding to the SA-magnetic beads. Triangles with asterisks indicate unique proteins found only in the presence of exogenously added bOVA and SA-magnetic beads. The arrow shows bOVA. (C) Microsomes with (+) or without (-) prior addition of bOVA were purified with (+) or without (-) SA-magnetic beads. Proteins (10 µg) or corresponding volumes of purified proteins were resolved on 7.5 – 15% SDS-PAGE, and subjected to Western blotting with SA-HRP. P.N.: post-nuclear fraction. Asterisks in the right indicate non-specific bands with SA-HRP. Equivalent results were attained by at least three independent assays. (D) Microsomes with prior addition of bOVA were purified with SA-magnetic beads. Microsomes were treated with (+) or without (-) trypsin and TX-100 before purification (left two lanes) or after purification (right two lanes). Proteins (2 µg) or corresponding volumes of purified proteins were resolved on 7.5 – 15% SDS-PAGE, and silver staining was used to visualize protein bands. Triangles on the right side indicate nonspecific proteins binding to the SA-magnetic beads. Triangles with asterisks indicate unique proteins found only in the presence of exogenously added bOVA and SA-magnetic beads. Equivalent results were attained by at least three independent assays. Reprinted with permission from reference28. Please click here to view a larger version of this figure.
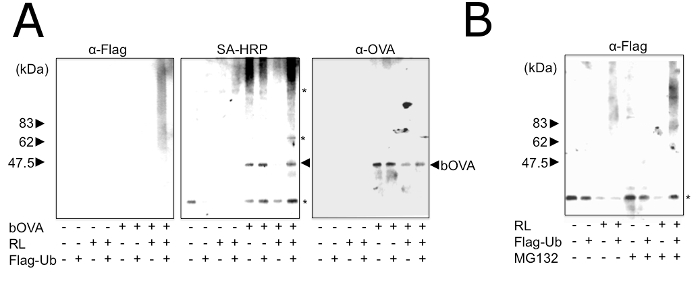
Figure 3: In Vitro Reconstitution of Processing and Ubiquitination using OVA in Purified Microsomes. (A) Purified microsomes with (+) or without (-) prior addition of bOVA were treated with (+) or without (-) RL and Flag-Ub for 1 h and were solubilized using TNE. bOVA was purified with SA-magnetic beads and subjected to Western blotting with the indicated antibodies. Asterisks in the right indicate non-specific bands with SA-HRP. Equivalent results were attained by at least three independent assays. (B) Purified microsomes were treated with (+) or without (-) RL, Flag-Ub, and MG132 for 1 h and were then solubilized using TNE. bOVA was purified with SA-magnetic beads and subjected to Western blotting with SA-Flag. Asterisks in the right indicate non-specific bands with SA-HRP. Equivalent results were attained by at least three independent assays. Reprinted with permission from reference28. Please click here to view a larger version of this figure.