The elucidation of the lncRNA interactome i.e., the cellular components that interact with lncRNAs, proteins, RNA, and DNA, is of key importance for understanding the functions of lncRNAs. Various techniques have been developed to characterize the lncRNA interactome, including RIP, CHART, ChIRP, and RNA pull-down. While the latter has been shown to be powerful in identifying RNA targets of lncRNAs, these procedures do not indicate whether the RNA partners interact indirectly via a protein network or directly via direct RNA/RNA interactions. However, it is now believed, that the mechanisms through which numerous lncRNAs exert their function rely on RNA/RNA interactions with other RNAs, often mRNAs, making crucial the development of methods for genome-wide discovery for these direct RNA targets. Furthermore, these RNA/RNA interactions, mediated by base pairing, can be cross-linked with psoralen derivatives6,5. Therefore, a procedure is proposed to identify RNAs engaged in direct RNA/RNA interactions with a lncRNA (workflow is depicted in Figure 1) and is based on the psoralen cross-linking of the cells coupled with RNA pull-down protocol.
The secondary structure of lncRNAs makes certain regions of lncRNA, particularly those with a low probability of internal base pairing, more capable of being hybridized by specific oligonucleotide probes than others. Then, as a first step of the procedure, the secondary structure of the lncRNA is modeled using the RNAstructure software7 to design oligonucleotides that target accessible regions and can hybridize specifically to lncRNA. Cells were lysed after cross-linking with psoralen and extracts were sonicated to obtain RNA fragments of ~2,000 nucleotides (Figure 1). Due to the length of some lncRNAs, this step is indispensable for their effective pull-down. Sonication of lncRNA generated short fragments that were pulled down using specific antisense probe located up to 2,000 nt upstream and 2,000 nt downstream from the probe binding site (Figure 1). Consequently, fragments corresponding to around 4,000 nt of a lncRNA were pulled with a specific probe (Figure 1). Then, high-throughput RNA sequencing or qPCR analysis can be performed after the RNA pull-down method to obtain the comprehensive list of RNA targets for a lncRNA of interest.
The procedure was applied to the lncRNA, nuclear-enriched abundant transcript one (Neat1). This lncRNA is essential to the formation and structure of nuclear bodies, the paraspeckles, found in the nucleus of the most cultured cells. In addition to Neat1, the paraspeckles contain several RNA binding proteins (RBP)14, and they retain RNA targets within the nucleus15. The nuclear retention of RNA targets by these nuclear bodies, i.e., paraspeckles, may occur through their binding to RBP or directly through their binding to Neat1 utilizing RNA/RNA interactions. To identify the RNAs targeted directly by Neat1 in a rat pituitary cell line, i.e., GH4C1 cells, the procedure whose graphical representation is given in Figure 1 was applied to the pull-down of Neat1 (Figure 2). In order to ensure the pull-down of the entire Neat1 long isoform (21 kb in length in rat), six Neat1-specific biotinylated probes (a-f, Table 1) that can bind to six different Neat1 parts were designed (Figure 2A). The parts of Neat1 pulled down by each probe are shown in Figure 2A. qPCR with specific primers targeting each of the 6 parts of Neat1 (Table 1), showed that each probe (a-f) was specific to the part of Neat1 against which it was designed (see Figure 2B).
In order to pull down the entire length of Neat1, the six probes (a-f) were pooled and named as pool SP1. To assess the specificity of the Neat1 RNA pull-down experiments, another additional pool of six probes (SP2) that were each designed in adjacent corresponding regions of binding of the six a-f probes, was used (Table 1). Every six parts of Neat1 were shown to be effectively pulled down with the SP1 pool, and the SP2 pool of the probes. However, the degree of recovery of each part was not the same (Figure 3A). Furthermore, the use of a non-specific probe (NSP, Table 1) recovered very low Neat1 as compared to the two specific pools SP1 and SP2 (Figure 3A).
To obtain a comprehensive list of direct RNA targets of Neat1 in GH4C1 cells, RNA-sequencing analysis was performed after Neat1 pull-down using the two specific probe pools described above16. It should be mentioned that low concentrations of RNAs recovered after Neat1 pull-down with NSP did not allow for the construction of libraries. Therefore, the lists obtained with the two specific probe pools directed to Neat1 (Table 1) were cross checked to assess the specificity of the results. 1791 RNAs were common in the two pools and corresponds to 70% of RNAs obtained with pool SP1 and 75% of RNAs obtained with pool SP2, respectively16. Some of these mRNAs were assessed with qPCR analysis using specific primers (Table 1). Consistent with results obtained in RNA-sequencing analysis, the transcripts Cntn1, Malat1, Pou1f1 and Rplp0 were found to be directly associated with Neat1 in RT-qPCR experiments (Figure 3B). By contrast, Tapbp a transcript not included in the list of direct RNA targets as determined by RNA-Seq, was not found enriched after RT-qPCR analysis (Figure 3B). The pull-down procedure described here has, therefore, proven to be an effective tool to explore the direct base-paired interactions between Neat1 and its RNA targets.
In addition, by using the 6 probes a-f separately, it was possible to assess the unique regions of Neat1 that were involved in RNA/RNA binding. Indeed, qPCR was performed for the five transcripts mentioned above using each of the 6 probes separately and compared to the non-specific probe (Figure 4). Each value of enrichment was normalized with the corresponding Neat 1 fragment percentage recovery. This was done to ensure that results obtained were independent of the degree of recovery for each Neat1 fragment by its corresponding probe sequences. The 4 mRNAs of Cntn1, Malat1, Pou1f1 and Rplp0 were shown to bind to the Neat1 5' end (Neat1_part1 bound by probe a). On the other hand, Tapbp was never enriched by any of the 6 probes (Figure 4). The procedure allows for the identification of the 5' region of Neat1 as the preferred location for base-paired RNA interactions.
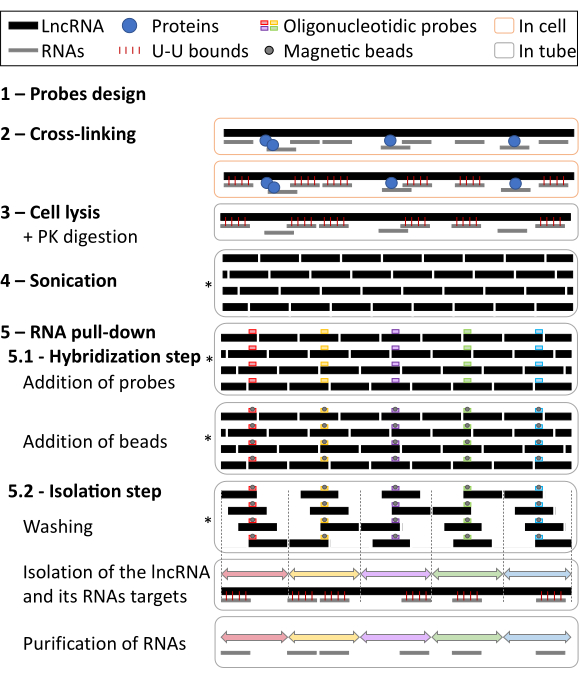
Figure 1: Graphical representation of the procedure. Cells are cross-linked with psoralen derivatives that intercalate into double-stranded RNA and, after irradiation with long-wavelength (365-nm), UV light covalently links pyrimidines on adjacent strands. Cells are then lysed, and proteins digested by Proteinase K. Sonication fragments RNAs to around 2000 nt fragments. Biotinylated specific probes are added for the hybridization with the lncRNA. Magnetic streptavidin beads are then added to separate specific material from the rest of the cell lysate. Beads are then isolated by a magnet and washed several times. After recovery RNAs are purified and used for RT-qPCR or RNA-seq analysis. In the four steps marked with (*), RNAs linked by U-U bounds ( ) are omitted to simplify the graphical representation. Please click here to view a larger version of this figure.
) are omitted to simplify the graphical representation. Please click here to view a larger version of this figure.
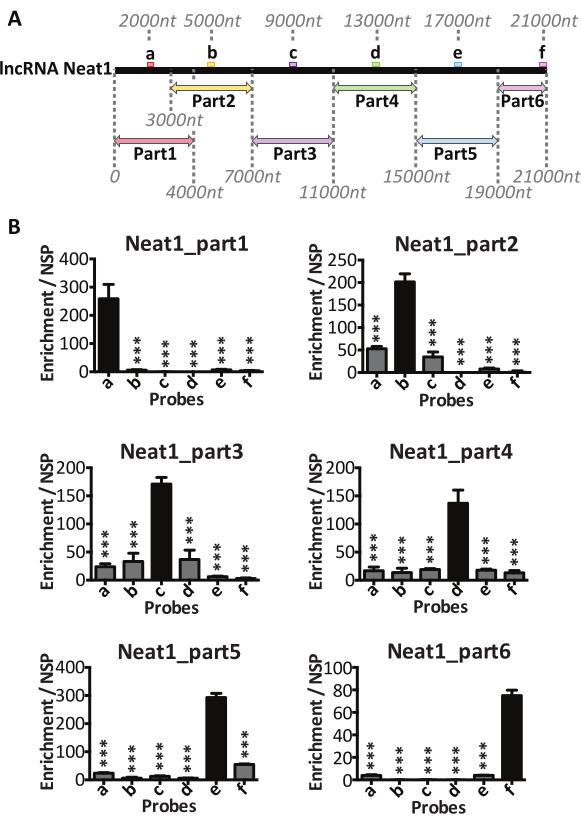
Figure 2: Pull-down of the different parts of Neat1. (A) Schematic representation of the pull-down of 6 parts of Neat1 by 6 specific probes (a-f). Schematic localization of the binding sites for 6 specific antisense oligonucleotides (a-f) designed along the length of Neat1. The different fragments of Neat1 generated by the sonication step, as well as the corresponding parts of Neat1 pulled-down by the different probes (a-f) are shown. (B) This figure has been modified from Jacq et al, 20219 reprinted by permission of Taylor & Francis Ltd, http://www.tandfonline.com. Specific parts of Neat1 pulled-down by each specific probe (a-f). The specific enrichment of the different parts of Neat1 was determined by RT-qPCR after pull-down with each specific probe (a-f) compared to a non-specific probe (NSP).*** p<0.0001 compared to the probe that specifically binds the part of Neat1 pull-down. Please click here to view a larger version of this figure.
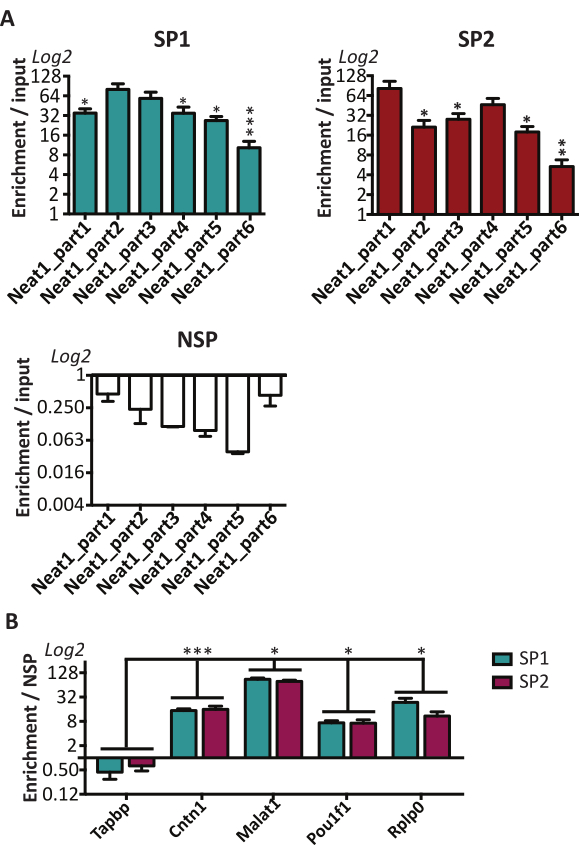
Figure 3: RNAs in direct RNA/RNA interaction with Neat1 identified by RT-qPCR. (A) This figure has been modified from Jacq et al, 20219 reprinted by permission of Taylor & Francis Ltd, http://www.tandfonline.com. The two pools of 6 specific probes, SP1 and SP2, were shown to induce a specific enrichment in every part of Neat1 as compared to a non-specific probe (NSP). However, while every 6 parts of Neat1 were efficiently pulled down both with the pool SP1 and the pool SP2 probes, the efficacy of both pools could differ depending on the part of Neat1 considered. SP1: *p<0.05 ***p<0.0001 compared to the Neat1_part2. SP2: *p<0.05 **p<0.01 compared to the Neat1_part1. (B) Four RNAs selected from the list of direct RNA targets established by RNA-seq were shown to be significantly enriched after Neat1 RNA pull-down with the pool SP1 and SP2 of specific probes relative to a non-specific probe (NSP) in contrast to a non-target RNA. *p<0.05 ***p<0.0001 compared to the non-target RNA (Tapbp). Please click here to view a larger version of this figure.
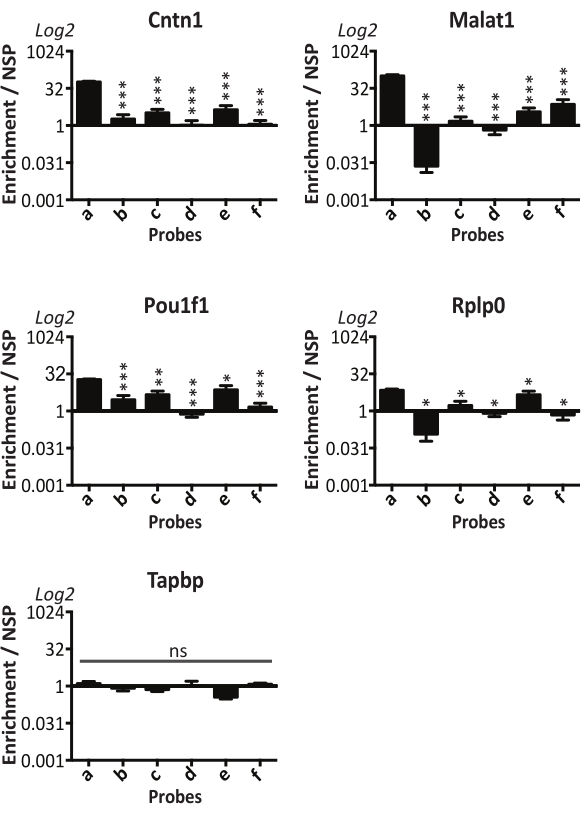
Figure 4: 5' region of Neat1 as the preferential location of base-paired RNA interactions. Each Neat1 specific probe (a-f) is used separately in RNA pull-down experiment and compared to a non-specific probe (NSP). Four Neat1 target RNAs (Cntn1, Malat1, Pou1f1 and Rplp0), as well as a non-target RNA (Tapbp) were analyzed by RT-qPCR. Enrichments relative to the NSP were normalized to the relative amount of corresponding Neat1_part captured. *p<0.05, **p<0.01, ***p<0.0001 compared to the probe a. Please click here to view a larger version of this figure.
| PROBE NAMES | SEQUENCES | |
| Specific Pool 1 (SP1) | ||
| a | CTCCACCATCATCAATCCTCTGGAC | |
| b | CATATAGCGGATGCCCAGGAACAAA | |
| c | ACAAAACAGAGCCCGAGAGTCAGTC | |
| d | ATGTACGTGACACGCTGACAACTGC | |
| e | ACTCCAAGATCTGACACCCTCACAC | |
| f | ACAGGGTCAGATGCAGTAAGACCTA | |
| Specific Pool 2 (SP2) | ||
| a | GCCTTCCCACATTTAAAAACACAAC | |
| b | TCCCCACAAGCATCTAAGAC | |
| c | TTACAATTACATACAGCCCTGTCCA | |
| d | AGTTGGTGAGTCCTAGCTCT | |
| e | TTAACCTTCCCTGGCAGTGT | |
| f | CAGGGTACTGCCTTGGTTTGGAAAT | |
| Specific Pool 2 (SP2) | ATAATTTCAAACATCAAATGGTATTTTA | |
| qPCR PRIMERS | FWD PRIMER | REV PRIMER |
| Neat1 Part_1 | AAGGCACGAGTTAGCCGCAAAT | TGTGCACAGTCAGACCTGTCATTC |
| Neat1 Part_2 | GCCTGCTTTCAGCTGTTGGTTT | TCTGGACAGCAACTGAGCAATACG |
| Neat1 Part_3 | TGCCAGTACTTCTGTCCCTAGGACAT | CTTCAGGCCTGGCTTCATTAGTGTTC |
| Neat1 Part_4 | GCTTTGGTGTATGGCGTGAGGTAA | CAGCCGACTGGGCAAAGCAATTAT |
| Neat1 Part_5 | GCTGACGTAGACTTTGAGGACCTACA | GAGATCGCTTGGGACCAGTTGGATAA |
| Neat1 Part_6 | GCATGAAGTTGGAAGCTGAGGGAAA | CTCTGAGACAGGGTCAGATGCAGTAA |
| Tapbp | GCAATTCCTGGGCTGCTTGAGAAA | TGTCCTTGCAGATAGGGCAGATGT |
| Cntn1 | GAGGTCTGGCTCCCGATACATAATCA | TGGGGACTTCTATGGAGTGCTTGT |
| Malat1 | GAAGGCGTGTACTGCTATGCTGTT | TCTCCTGAGGTGACTGTGAACCAA |
| Pou1f1 | TTCCAGAGGATGTGGGTTCCATCA | CTGTGCTCCATTTTCAGGCCAAGT |
| Rplp0 | CTTCCCACTGGCTGAAAAGGTCAA | AAGAGACCGAATCCCATGTCCTCA |
Table 1: Sequences of DNA oligonucleotide probes and qPCR primers
