Following the protocol (Figure 1A), hiPSCs were differentiated into MSCs via the EB formation and monolayer culture methods. During differentiation, the cells showed different representative morphologies (Figure 1B,C).
As shown in Figure 1B, the hiPSCs colonies display typical compact morphology before differentiation with a clear border composed of tightly packed cells. Uniform spherical EBs formed after hiPSCs dissociating and culturing for 24 h on the shaker. During day 1 to day 7 of culture in MSCs differentiation medium, the smooth edge of EB became rough, and the volume of EB grew big. From day 8 to day 17, after transferring the EBs to a GFR-extracellular matrix gel-coated 6-well plate, EBs gradually adhered to the plate, and many adherent monolayer cells spread around the EBs. When the cells reached 90% confluency on day 18, the cells were digested and seeded on a gelatin-coated culture plate. On day 19, the cell adhered and showed a polygonal shape. Consecutively, the cells were passaged twice when they were 90% confluent. The derived MSCs gradually matured and showed a typical spindle shape, and the colony grew in a swirl.
As shown in Figure 1C, the volume of the cells increased and spread around the colony after replacing the iPSC maintenance medium with the MSC maintenance medium for 24 h. While cultured in an MSC maintenance medium, the cells gradually proliferated and formed multilayer adherent cells. On day 14, the cells were digested and seeded on a gelatin-coated culture plate. On day 15, the cell adhered and showed a polygonal shape. Consecutively, the cells were passaged 6 times when they were 90% confluent. The derived MSCs gradually matured and showed a typical spindle shape, and the colony grew in a swirl.
The surface antigens of hiPSCs and hiPSC-driving MSCs were analyzed by flow cytometry (Figure 2). As shown in Figure 2C, hiPSCs were positive for CD90 and negative for CD34, CD45, CD73, and CD105. After differentiating hiPSCs into MSCs via both methods, driving MSCs were positive for CD90, CD73 and CD105, and negative for CD34, and CD45 (Figure 2A,B).
The differentiation capability of hiPSC-driving MSCs was investigated by osteogenic, adipogenic, and chondrogenic differentiation. As shown in Figure 3, both hiPSC-driving MSCs of the two methods differentiated into osteoblast, adipocyte, and chondrocyte. The hiPSC-driving MSCs of the monolayer method formed more calcium deposits than the EB method (Figure 3B). The two methods had no significant difference in adipogenic differentiation and chondrogenic differentiation ability (Figure 3D,F).
The proliferation ability of hiPSC-driving MSCs was examined by continuous passage culture. As shown in Figure 3G, the hiPSC-driving MSCs of both methods can be passaged for more than 20 passages and still maintain a rapid proliferation ability.
The comparison between the two approaches shown in Table 1 is based on differentiation time, cost, cell proliferation ability, MSC markers' expression, and their differentiation capability in vitro.
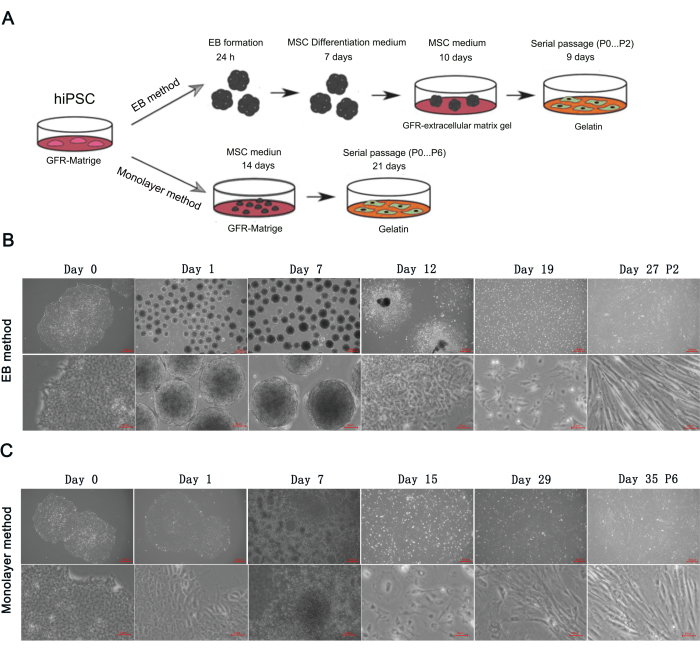
Figure 1: Differentiating hiPSCs into MSCs via EB formation method and monolayer culture method. (A) Schematic showing the differentiation of hiPSCs into MSCs via EB formation and monolayer culture. Representation morphology of cells at key phages during MSCs derivation from hiPSCs via (B) EB formation and (C) monolayer culture. Scale bars: 300 µm and 50 µm. Please click here to view a larger version of this figure.
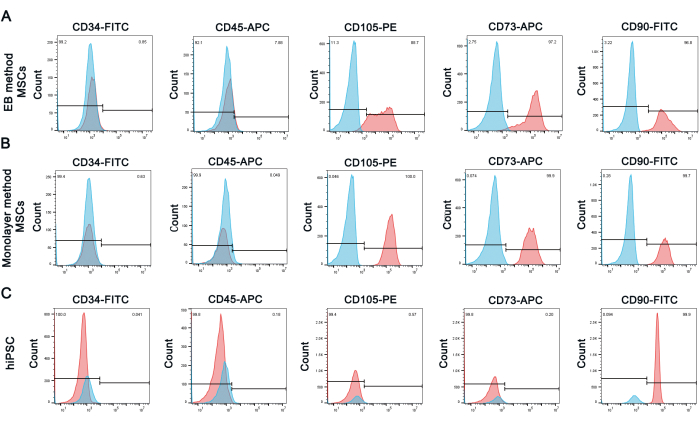
Figure 2: hiPSC-driving MSCs surface antigens analysis by flow cytometry. Expression percentages of MSC surface antigens in iPSC-driving MSCs via (A) EB formation and (B) monolayer culture. (C) hiPSCs were used as the negative control. MSCs negative markers: CD34, CD45; MSCs positive markers: CD73, CD90, and CD105. hiPSCs negative markers: CD34, CD45, CD73 and CD105; hiPSCs positive markers: CD90. Please click here to view a larger version of this figure.
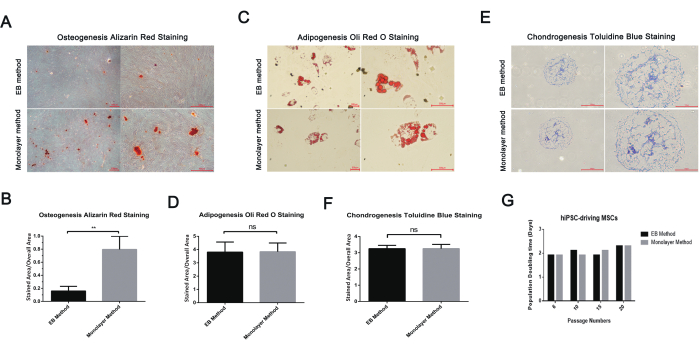
Figure 3: Three-line differentiation and proliferation ability of hiPSC-driving MSCs. (A) Alizarin red staining of calcium deposits of hiPSC-driving MSCs in osteogenic differentiation medium for 2 weeks. Scale bars: 300 µm. (B) Quantification of Alizarin red S staining by ImageJ analysis. (C) Oli red O staining of lipid droplets of hiPSC-driving MSCs in adipogenic differentiation medium for 2 weeks. Scale bars: 300 µm. (D) Quantification of Oli Red O staining by ImageJ analysis. (E) Toluidine blue staining of extracellular chondrocyte matrix. Scale bars: 300 µm and 150 µm. (F) Quantification of toluidine blue staining by ImageJ analysis. (G) hiPSC-driving MSCs population doubling time calculation. Please click here to view a larger version of this figure.
| Comparison | EB formation method | Monolayer cultures method |
| Differentiation time | 27 days | 35 days |
| Cost | High | Low |
| Proliferation Speed | Fast | Fast |
| Proliferation ability | ≥20 passage | ≥20 passage |
| Expression of MSC markers | CD73/CD90/CD105 positive,CD34/CD45 nagative | CD73/CD90/CD105 positive,CD34/CD45 nagative |
| Capability of differentiation | Adipogenic differentiation, chondrogenic differentiation and osteogenic differentiation capability | Adipogenic differentiation, chondrogenic differentiation and stronger osteogenic differentiation capability |
| Operation | Complicated | Simple |
Table 1: Characteristics of the two methods for differentiating hiPSCs into MSCs.
