This protocol describes a method to track experience-dependent molecular changes in individual cortical neurons in live animals. A chronic cranial window is first created over a cortical region of interest in a mouse carrying a fluorescent reporter of gene expression. Two-photon microscopy can then be coupled with various behavioral paradigms to observe behaviorally induced molecular changes in individual neurons and track such changes in the same sets of neurons over multiple days (Figure 1).
In Arc-GFP mice, experience-dependent changes in Arc gene expression can be reliably imaged in individual cortical neurons for multiple days using this protocol. The transgenic Arc-GFP knock-in mouse expresses a destabilized green fluorescent protein (d2EGFP, protein half-life approximately two hours) under the control of the endogenous promoter of the immediate early gene Arc (Figure 2).
Past protocols describing chronic cranial windows have outlined critical steps in conducting successful cranial window surgeries11. This protocol provides additional guidance in removing the dura if needed, which increases the likelihood of obtaining clear optical windows when such windows are situated over skull sutures (see Step 2.4).
After recovery from cranial window surgery, animals are mounted in a custom-made head-fixation frame and stage. Figure 3 depicts the headbar and head-fixation frame used during two-photon imaging. Maintaining a consistent two-photon microscope imaging stage position and setup will facilitate day-to-day realignment of images when returning to a previously imaged brain region, and increase experimental efficiency when multiple animals are imaged on the same day (Figure 4).
During behavioral training or environmental stimulation, it is advisable to house animals singly, and in an environmentally consistent home cage location, to minimize day-to-day fluctuations in background Arc-GFP activation levels (Figure 1).
A critical sign of a stable, clear cranial window is the crisp pattern of blood vessels under epi-fluorescent illumination. This blood vessel pattern should not change significantly across multiple days of imaging (Figure 5).
Figure 6 illustrates the expression patterns of Arc-GFP in frontal cortical neurons under a baseline home cage condition (A) and after performance of a new motor behavior (B). Layer II/III cortical neurons in the same brain region in the same animal can be reliably imaged, and the same neurons should be able to be identified over multiple days of imaging. It is typical to collect a 3-D image stack to sample hundreds of neurons. Neuron edges should be sharp and clear throughout the image stack. Tissue auto-fluorescence in the red channel also provides an index of window clarity. If the imaged region becomes drastically murkier over days, the cranial window quality is likely deteriorated. A typical cranial window will remain optically clear for at least a week. After several weeks to months, the regrowth of skull from the edges of the cranial window and thickening of the dura will eventually prevent further imaging experiments.
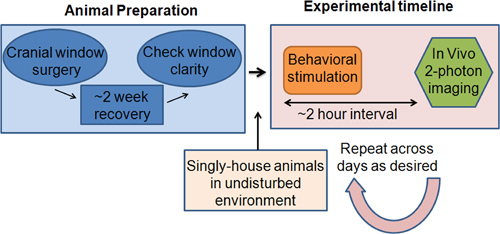
Figure 1. A schema outlining the experimental steps. During the Animal Preparation phase, cranial window surgeries are performed on Arc-GFP mice. The animals are given about two weeks to recover from the surgery in their home cage. The clarity of the cranial windows is then checked using epi-fluorescence microscopy. If the windows are ready for imaging, the animals are singly housed in a quiet, environmentally consistent home cage environment, whence the Experimental Timeline phase can begin. The behavioral stimulation protocol and the imaging interval may be tailored to detect Arc-GFP at its peak fluorescence, which typically occurs two hours after activation. After the behavioral stimulation is complete, the animal is anesthetized and the cortical region of interest is imaged through the cranial window using two-photon microscopy. The animal is then returned to the home cage. The behavioral and imaging sessions can be repeated across days as desired.

Figure 2. A diagram illustrating the construction of the Arc-GFP knock-in mouse line, in which a gene encoding destabilized green fluorescent protein replaced the coding part of the immediate early gene Arc. In this strain of mouse, GFP expression is under the control of the endogenous Arc promoter. This diagram is redrawn based on the description in Reference 10.
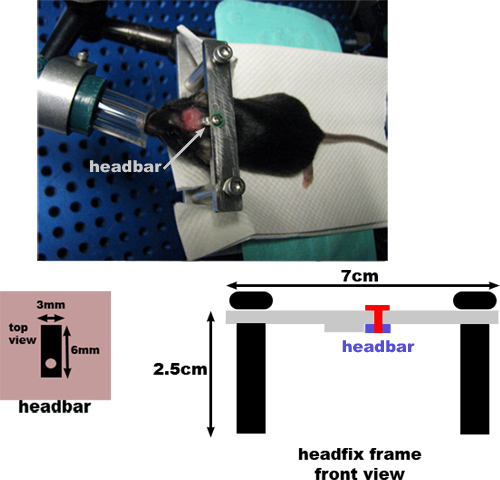
Figure 3. Schematic and dimensions of the head-fixation setup used for two-photon imaging. The solid half of the headbar is glued to the back of the skull (Section 2.8), and the end with the screw hole is used to attach the anesthetized animal’s head to the frame. The head-fixation frame consists of a thin metal plate mounted atop two poles with screws.

Figure 4. Example of a laser scanning two-photon microscope setup for Arc-GFP mice imaging. Note the 25x water immersion lens, heating pad and custom microscope stage with a head-fixation frame. The anesthetized Arc-GFP mouse shown here is ready for in vivo imaging.
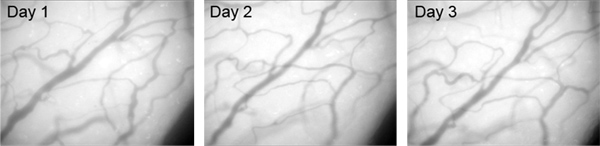
Figure 5. Images of brain surface blood vessels in a chronic cranial window over multiple days, showing the clarity and stability of blood vessel patterns over time. Brain surface was illuminated with blue light, and the images were taken through a 25x water immersion lens by a CCD camera mounted on the microscope.
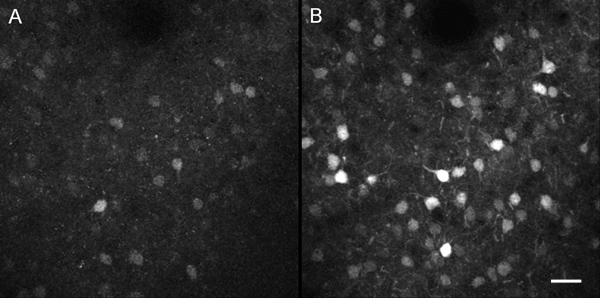
Figure 6. In vivo two-photon images of Arc-GFP expression patterns in frontal cortical neurons of a mouse after two different behavioral experiences. (A) The mouse stayed in its home cage before imaging on the first day. (B) The mouse performed a new motor behavior before imaging on the second day. Fluorescent images of the same cortical region were simultaneously acquired in green and red channels. GFP fluorescence only appeared in the green channel, whereas tissue autofluorescence appeared in both channels. Detection parameters were set so that tissue auto-fluorescence levels in both red and green channels had equal readings, and these parameters were maintained throughout experiments. The red channel signal was then subtracted from the green channel signal to remove broad-band tissue autofluorescence during off-line analysis. The resulting green fluorescent signals from an 18 μm thick 3-D image stack were projected by maximum intensity to the horizontal plane, to provide a top-down view of the cellular patterns of Arc-GFP expression. Scale bar, 30 μm.
