Figure 1 shows a representative result of the Nucleocapsid Annealing Mediated Electrophoresis (NAME) assay, performed i) with the full-length recombinant protein, ii) without the protein, i.e. mixing cTAR + TAR and incubating up to 1 hr at RT, and iii) the same assay performed with (12-55)NC, a synthetic peptide lacking the 11 amino acids of N-terminal domain. The pre-folded TAR and cTAR were mixed and the formation of the annealed heteroduplex TAR/cTAR was monitored in the presence of the full-length recombinant NC (left lanes of Figure 1), in the absence of protein (central lanes in Figure 1), and in the presence of the truncated (12-55)NC peptide (right lanes in Figure 1). Controls are: folded cTAR, folded TAR, and annealed hybrid (TAR/cTAR), obtained through thermal denaturation of the stably folded structures of TAR to cTAR followed by their slow annealing.14
The ability of NC to denature two stable nucleic acid sequences into the extended heteroduplex helix is clearly evident: the full-length NC protein leads immediately to the formation of the TAR/cTAR hybrid: 0’ indicates the minimal time passing between addition of NC to the samples and addition of gel loading buffer, which stops the reaction; complete formation is already achieved after 15’ incubation time. The increase in the intensity of the annealed heteroduplex parallels the decrease in the intensity of the folded cTAR and TAR oligonucleotides. The heteroduplex formation is achieved also in the presence of the truncated form, i.e. the (12-55)NC, confirming the biological activity of the synthetic peptide. In the absence of the protein or of the peptide the heteroduplex formation is not observed, and TAR and cTAR oligonucleotides will not anneal at room temperature into the heteroduplex (Figure 1). In all experiments, the fast formation of unstable intermediates (“intermediate complexes” in Figure 1) that decrease over time is observed, consistently with the mechanism proposed of strand exchange through the hairpins of cTAR and TAR till the correct nucleic acids folding.17
A possible troubleshooting of the experiment is the lack of SDS in the Gel Loading Buffer. In the absence of SDS in the GLB the outcome of the reaction is shown in Figure 2 and compared to the outcome with GLB + SDS. The pre-folded TAR and cTAR were mixed and incubated for 3 hr at RT of for 3 hr at 37 °C with the recombinant full-length NC protein. After incubation the samples were split in two: to one half gel loading buffer without SDS (GLB) was added, while to the other half GLB was with SDS (GLBSDS). Samples were loaded on the gel, and the position of the nucleic acids bands was visualized after the gel run by dye staining. When NC was present but the samples were loaded with GLB, all nucleic acids bands were shifted up: this is expected and indicates a strong binding between the protein and the nucleic acids. The results with GLBSDS are shown in the extreme right of the gel: the addition of SDS denatures the protein and releases the nucleic acids from the stable complex with the protein, allowing the comparison with the controls.
The NAME assay is used to assess the ability of threading intercalators to stabilize dynamic nucleic acid structures and inhibit NC chaperone activity.14 Threading intercalators are planar aromatic molecules substituted by bulky side chains located at the opposite sites of the ring system, such as the anthraquinone shown in Figure 3A.18 These intercalators are able to thread through the double helix of nucleic acids, finally locating their side chains in each groove of the double helix.19,20
Figure 3B shows the outcome of the NAME assay in the presence of compound 1, a known threading intercalator,18 in the presence of full-length NC or of the truncated peptide. In both cases, the formation of the TAR/cTAR hybrid by NC is reduced by increasing the concentration of the intercalator, while, at the same time, the amount of free TAR and cTAR increases. The truncated form of the protein lacking the N-terminal tail (12-55NC) is more sensitive to the inhibition of its activity: this difference was verified with all the compounds tested so far.
The NAME assay can be performed in two ways, either by pre-incubating the test compound with NC, followed by addition of the folded oligonucleotides (NC-preincubation mode), or by preincubating the test compound for 15 min with the folded nucleic acid substrates, followed by addition of NC and a further incubation for 15 min (oligo-preincubation mode). Although the overall incubation time is the same in the two different modes, the preincubation with the folded oligonucleotides before addition of the NC allows more time for the threading of the intercalators into the folded nucleic acid. This results in a stronger stabilization of their dynamic structures and into an easier inhibition of NC chaperone activities. The analysis of the NAME assay in the two modes therefore enhances differences in the mechanism of action of NC inhibitors, as shown in Figure 4: when compound 1 was analyzed by NAME in the oligo-preincubation mode (Figure 4, lanes at the left) it shows potent inhibition of NC-mediated annealing, whereas much lower inhibition is observed in the NC-preincubation mode (Figure 4, lanes at the right). Please note that for the determination of inhibitory concentrations while screening sets of related compounds with this assay and in these conditions, each experiment must be performed at least in triplicate using closely spaced concentrations (especially around the IC50 value).14
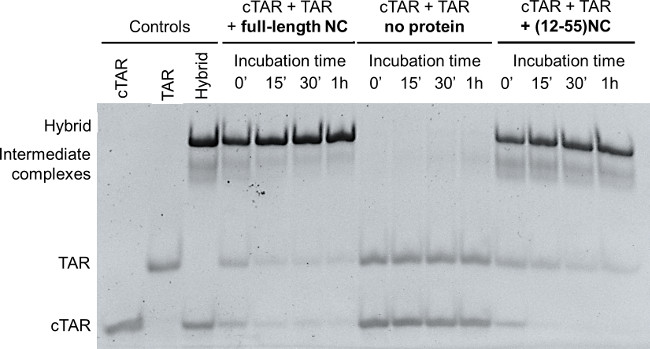
Figure 1: NAME assay with full-length NC and with the truncated (12-55)NC. Folded TAR and cTAR, each 1 µM, were incubated with recombinant NC or with (12-55)NC (oligos/NC=1/8) for 0 min, 15 min, 30 min and 1 hr. TAR and cTAR incubated in the absence of protein (0 min, 15 min, 30 min and 1 hr) were used as negative control. Folded TAR, folded cTAR and the hybrid TAR/cTAR obtained after thermal denaturation followed by in vitro annealing were used as controls. The reactions were then stopped using GLBSDS. Samples were resolved on a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator.
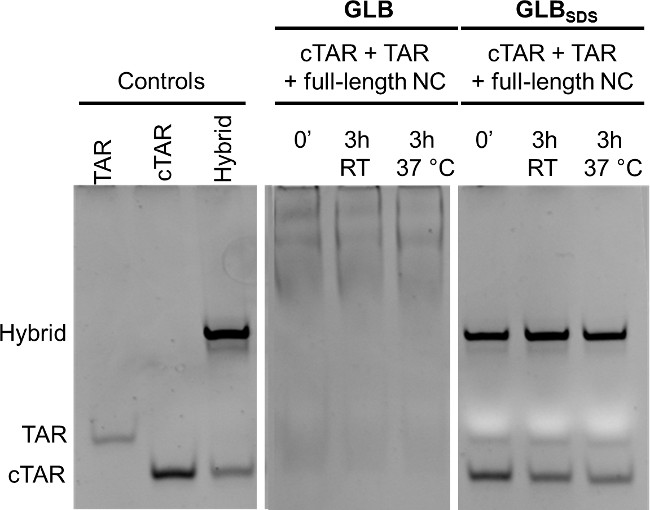
Figure 2: Effect of SDS in gel loading buffer (GLB) when analyzing TAR/cTAR annealing by NC. TAR and cTAR, prefolded in TNMg 1x, were incubated with full-length recombinant NC (oligos/NC = 1/8) for 3 hr at room temperature or at 37 °C. GLB or GLBSDS were then added to the samples incubated with NC. Controls: TAR, cTAR and the annealed hybrid TAR/cTAR (each 1 µM). Electrophoresis was performed using a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator. This figure has been modified from Figure S4 of: Sosic, A. et al. Design, synthesis and biological evaluation of TAR and cTAR binders as HIV-1 nucleocapsid inhibitors. MedChemComm 4, 1388-1393, doi:10.1039/c3md00212h (2013).
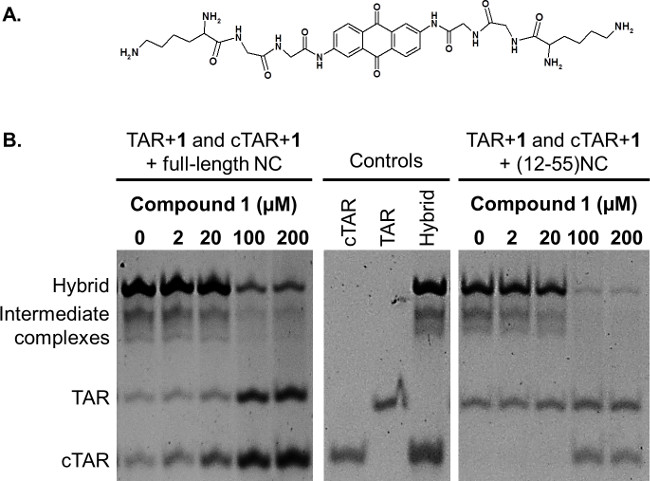
Figure 3: (A) Chemical structure of the threading intercalator 1. (B) Effect of threading intercalator 1 assessed by NAME assay. Folded TAR and cTAR, each 1 µM, were incubated in the presence of the indicated concentrations of the compound 1 with the full-length NC or with the (12-55)NC, each 8 µM. Folded TAR, folded cTAR and the extended heteroduplex TAR/cTAR were used as controls. The reactions were then stopped using GLBSDS. Samples were resolved on a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator.
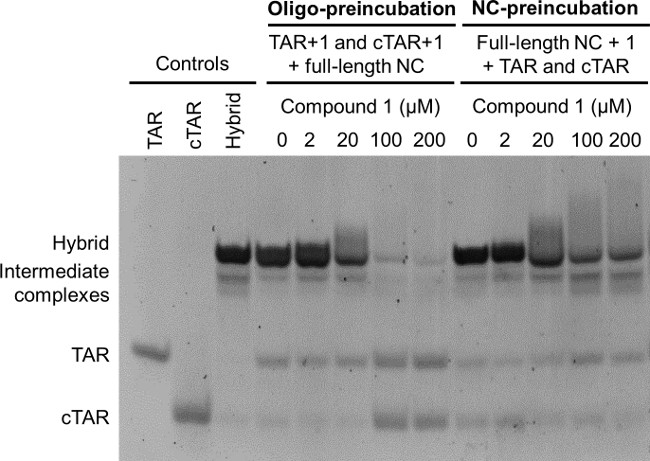
Figure 4: Oligo-preincubation versus NC-preincubation modes: effects on NAME assay. Oligo-preincubation mode: folded TAR and folded cTAR, each 1 µM, were preincubated with the indicated concentrations of the threading intercalator 1 for 15 min, and then for additional 15 min in the presence of the full-length NC (8 µM). NC-preincubation mode: the indicated concentrations of the threading intercalator 1 were preincubated with the full-length NC (8 µM) for 15 min, and then for additional 15 min in the presence of folded TAR and folded cTAR, each 1 µM. Folded TAR, folded cTAR and the extended heteroduplex TAR/cTAR were used as controls. All reactions were finally stopped using GLBSDS. Samples were resolved on a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator.
