固体と液体の腫瘍におけるアクショナブル変異の検出のための次世代シーケンシング
Summary
This manuscript describes clinical protocols for two next-generation sequencing panels. One panel interrogates hematologic malignancies while the other panel targets genes commonly mutated in solid tumors. Molecular classification of driver mutations in human malignancies offers valuable prognostic and predictive information.
Abstract
As our understanding of the driver mutations necessary for initiation and progression of cancers improves, we gain critical information on how specific molecular profiles of a tumor may predict responsiveness to therapeutic agents or provide knowledge about prognosis. At our institution a tumor genotyping program was established as part of routine clinical care, screening both hematologic and solid tumors for a wide spectrum of mutations using two next-generation sequencing (NGS) panels: a custom, 33 gene hematological malignancies panel for use with peripheral blood and bone marrow, and a commercially produced solid tumor panel for use with formalin-fixed paraffin-embedded tissue that targets 47 genes commonly mutated in cancer. Our workflow includes a pathologist review of the biopsy to ensure there is adequate amount of tumor for the assay followed by customized DNA extraction is performed on the specimen. Quality control of the specimen includes steps for quantity, quality and integrity and only after the extracted DNA passes these metrics an amplicon library is generated and sequenced. The resulting data is analyzed through an in-house bioinformatics pipeline and the variants are reviewed and interpreted for pathogenicity. Here we provide a snapshot of the utility of each panel using two clinical cases to provide insight into how a well-designed NGS workflow can contribute to optimizing clinical outcomes.
Introduction
臨床腫瘍学の標本の次世代シーケンシング(NGS)は、ターゲッティング遺伝的変化および予測/予後分子マーカーを同定することの重要性に科学文献点を成長させるように、過去数年間でより広く利用可能になりました。マルチ遺伝子パネルは、分析し、病気が進行し、再発の両方の上皮1,2および血液3悪性腫瘍における全体exomeシーケンシング研究では、腫瘍の不均一性およびクローン進化の概念を固めてきました。さらに、そのようなポリメラーゼ連鎖反応(PCR)またはサンガー配列決定のような競合する技術とは異なり、NGSは、単一のアッセイ4内のすべての臨床的に関連する癌遺伝子における最もゲノム変化を検出することができます。
当初は2臨床NGSパネル、カスタム血液学パネル(ヘム-NGSパネル)および既製FFPE標本のためのがんパネル(ソリッドNGSパネル)で起動パーソナライズ診断センター(参照<strong>図1)。これらのパネルは、選択された遺伝子の臨床的に関連または高い関心領域をカバーします。すべての遺伝子またはエキソンが完全に覆われていません。アンプリコンを延長し、ライゲーションに続いてプローブハイブリダイゼーションによって生成されます。対象地域はさらに、配列決定のためにプールされる96サンプルまで可能、ユニバーサルデュアルインデックスプライマーを用いてPCRを用いて増幅しています。
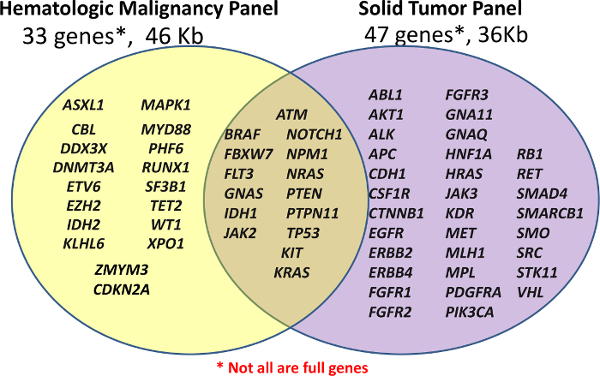
図1:パネルで覆われた遺伝子のリストライブラリ調製は33の遺伝子または既製のアンプリコンがんパネル(ソリッド-NGS)47遺伝子ののカスタム血液学パネル(ヘム-NGSパネル)のいずれかを使用して行われます。いくつかのアンプリコンは唯一の特定のホットスポットをカバーすることができるようにすべてではない遺伝子またはエキソンのは、完全に覆われている。 この図の拡大版をご覧になるにはこちらをクリックしてください。</ A>
ヘム-NGSパネルのコンテンツは、複数のソースから派生しますが、以前の臨床的有用性5の高さを実証するものとして説明急性骨髄性白血病(AML)に変異した16の遺伝子を中心としました。ソリッドNGSパネルは、商業的に(COSMIC)データベース6癌における体細胞突然変異のカタログで報告されているように癌で一般的に変異した遺伝子に基づいて対象地域で生産されています。
いくつかの重要なステップは、臨床NGSのためのワークフロー全体を特徴づけます。臨床医が検査を注文した後、病理医は腫瘍の割合とサンプルボリュームの分析を以下の試料の妥当性を判断します。我々の施設では、技術の背景配列決定エラー率(「騒音」)と標的アプローチの効率のために、少なくとも10%の腫瘍を必要とします。組織検査のために十分である場合には、ゲノムDNAを抽出します。このDNAは、次いで、(複数の品質管理にかけられますQC)の手順。 DNAは、QCに合格した場合、アンプリコンライブラリーが生成され、配列決定されます。得られたデータを、社内のバイオインフォマティクスパイプラインを介して分析されます。バイオインフォマティクス解析に続いて、変異体は、手動で見直され、臨床報告に組み込む前に、病原性のために解釈しました。下に私たちは、この厳格なワークフローを経て、最終的には臨床管理の変化につながった2例を説明します。
ケース1 -急性骨髄性白血病
患者Aからの骨髄生検は成熟せず、AMLのための診断でした。細胞遺伝学的研究は、骨髄標本に送信され、正常な女性核型を示しました。 95%の循環芽細胞は、そのように、末梢血試料はヘム-NGSパネルのパーソナライズされた診断テストのために送られた、存在がありました。
急性骨髄性白血病(AML)は、白血球の骨髄系統の血液学的な悪性腫瘍です。検出AMLにおける遺伝子変異の病因と予後7に重要と認識再発性遺伝子の変異と、予後および治療 のためにますます重要になってきています。 FLT3内部タンデム重複(ITDS)が少なく良好な治療成績8に関連付けられてきたがNPM1とCEBPAの変異は、良好な予後のリスクと関連しています。証拠の成長体は、AML 9におけるこれらおよび他の変異のための病原性の役割をサポートしています。
ケース2 -肺腺癌
患者Bから左鎖骨上の質量の生検は、肺腺癌を実証しました。 50%を超える腫瘍を有するロール/カールは、変異が標的治療的介入のために存在していたかどうかを識別するためにとして、ホルマリン固定パラフィン包埋(FFPE)リンパ節塊から生検材料は、ゲノムテスト(ソリッドNGSパネル)のために送られました。
肺CANCERは、米国における癌関連死亡の主要な原因であり、2つの主要なタイプの非小細胞肺癌(NSCLC)および小細胞肺癌(SCLC)に分割されています。 NSCLCはさらに、病変の組織学に基づいて、腺癌または扁平上皮癌のいずれかとして定義することができます。肺腺癌は喫煙者と非喫煙者の両方で見られる肺癌の最も一般的なサブタイプであり、かつ非喫煙者10のための肺癌の最も一般的な形式です。肺腺癌の分子の研究では、複数の癌遺伝子11の変異を同定しました。喫煙者で同定された最も一般的なドライバの変異は、KRASおよびBRAFにおける突然変異です。非喫煙者の中で最も一般的な変異は、EGFRにおける変異、および遺伝子ALK、RETとROS1を伴う再編成されています。肺腫瘍は、遺伝子ERBB2(HER2 / neuの )におけるインフレームエクソン20挿入に記載されています。 HEで最も一般的な異常R2 / neuが標的治療は、(:HER2 / neuのに対するヒト化モノクローナル抗体トラスツズマブ)使用可能な乳癌におけるこの遺伝子座の増幅です。 2で観察されたHER2 / neuのエクソン 20挿入-肺の4%が12(それぞれ、ネラチニブとテムシロリムス)13 HER2 / neuおよびmTOR阻害剤との併用療法に部分的な応答を示しているadenocarcimomas。
Protocol
Representative Results
Discussion
この原稿で説明する2つのNGSの試験が臨床的に提供されているように、最も重要な実用的な考慮事項は、品質管理です。具体的には、密接な考慮事項は、抽出したDNAの質と量に支払わなければなりません。これは、多くの場合、変数のDNA収率で高度に分解されFFPEサンプルのために特に重要です。イソプロパノール沈殿法、カラムベースの方法は時々制限された溶出体積を有するDNAの剪断をもたらすことが見出されたようFFPEサンプルからのDNAの収量を最大化するために開発されました。したがって、試料が低すぎる濃度を与えるまたはアッセイのためにあまりにも低下している時間のほとんどは、それが原因で、組織の大きさ、種類、または固定ではなく、抽出プロセスに最も可能性が高いです。抽出の失敗がある場合、血液/骨髄標本のために、それが原因hemodilute又は化学切除( すなわち 、不十分その抽選で白血球又は腫瘍細胞の数を有する)であるサンプルに通常です。
DNAの品質が良好であればしかし、その後、低入力量は、成功することができ; 250 ngの多くの場合、アッセイで使用されている – NT ">検証中に、DNAの質と量の受容のためのカットオフは、100の推奨入力を確立すべきです。 DNAの品質( すなわち 、増幅可能なDNAの量が100未満である- 250 ngの)悪い場合に加えて、(増幅可能なDNAの量が推奨される入力に到達するため)、次に高い入力量は、シーケンシング結果の品質を向上させることができ。DNAの質と量のためのメトリクスは、ライブラリの準備にDNAを進める前に、各サンプルに適用する必要があります。これらのサンプルを「グレーゾーン」( 図2参照)には、実験室のディレクターまたは指名の裁量で実行する必要があります。現時点でのベストDNAは、塩基配列決定の際に十分に機能していないかどうかを予測する方法は、入力DNAの定量および品質評価が可能になる定量PCRベースのアッセイを行うことである。このアプローチは、bioavailabに対処します異なるサイズのフラグメント( 例えば、100塩基対、150塩基対、200 bpおよび300 bp)の比較収量の増幅を経て、検体中の異なるサイズのフラグメントのility。現在、ライブラリの準備にはいくつかの節目の1に失策が失敗した場合や低品質のものであるとのいずれかにライブラリを引き起こす可能性が手動手順が多数含まれます。マイクロ流体ゲル分析を、配列決定の前に、ライブラリの準備の問題をチェックするための唯一のQC工程です。したがって、余分なマインドフルネスは、反応の成功の確率を高めることができますいくつかの重要なステップがあります。正確なサンプルとオリゴヌクレオチドプールを、各試料のために使用されることを保証することが不可欠です。確実かつ適切に各サンプルは二重インデックスPCRプライマー対の96のユニークな組み合わせのいずれかがサンプルミックスアップするための機会を減少させる含まれていることを記録します。また、正しくフィルタープレート(FPU)ドレインを確保することが重要です。それが適切に排水しない場合、これはエクステを引き起こす可能性がありますライブラリの準備のnsion-ライゲーション工程は、次善行い、質の悪い配列決定データをリードします。ライブラリQC後、LNB1ビーズを完全に再懸濁し、LNB1 / LNA1溶液をこの混合物の濃度は、ライブラリーのモル濃度を決定するために使用されるように、サンプルに追加する前に十分に混合することをされていることを確認することが重要です。ビーズ溶出工程は、ビーズをオフに溶出するライブラリの次善の量につながる場合最後に、クラスタリングの密度を減少させ、場合によっては適切な平均カバレッジを取得しないように、ライブラリの原因となります。逆に、ライブラリーの過剰は、貧しい品質が読み込みにつながります。したがって、シーケンサ上のライブラリの最適なプーリングとクラスタリングを確保するためのビーズベースの正規化ステップで一致することが重要です。
ライブラリーの調製に加えて、生の、逆多重化FASTQファイルから正確な変異コールを生成するバイオインフォマティクスパイプラインを検証することが重要です。選択1を取捨選択しなければならない多くのオープンソースと市販のアライナ、バリアント発信者、およびNGSソフトウェアパッケージがあるので、カスタムソリューションは、時間がかかる場合があります。カスタムアルゴリズムは、本質的なパフォーマンス統計を抽出し、ほとんどのオープンソースツールを逃れたユニークな定期的な変異を同定し、遺伝子座の各々の上にコピー数の状態を決定するように設計する必要があります。バイオインフォマティクスパイプラインの検証プロセス中に、満たす、または品質のフィルタリング後のカバレッジの最小深さ( 例えば 、250の最小読み取り)と最小対立遺伝子頻度の両方を超える変異体についての報告義務のカットオフ値を決定することが重要である( 例えば 、4 %)。この多重化されたアンプリコンベースのアッセイので、カバレッジの最小平均深さ( 例えば 、1,000倍)ライブラリの読み込みの最小深さを最低実行アンプリコンを得ることができるようにするために達成する必要があることを決定することが重要です。また、検定の多重化の性質は、cを行いますauseオフターゲット効果とこれらの「成果物」を発見し、完全に起動する前に吟味する必要があります。記載されたアッセイの別の重要な制限は、検証最小対立遺伝子頻度を達成するために、10%を超える腫瘍を含有する試料について必要です。
低周波数の検出は、1%、FLT3の挿入は手動レビューがこの過程でまだ望ましいことの証拠です。でも5%の対立遺伝子頻度カットオフを有する、いくつかの重要な突然変異は多分逃したので、マニュアルレビューは、これらの変異体を確認することが不可欠となります。 FLT3-ITDSについては、エクソン14の目視検査は見過ごさない低レベルまたは大規模な挿入/複製を確保するために、すべてのAML患者に対して行われます。また、一般的にプライマー配列の隣にあるHER2エクソン20の挿入は、手動による介入が必要です。堅牢なバイオインフォマティクスパイプラインを持つにもかかわらず、いくつかの変異体は、ハードカットを持つだけの性質である見落とさ行くことができます上記のほとんどの統計のためにオフ。より良いライブラリーの調製および/または配列決定の方法論が意志として少ないアーティファクトや偽陽性が含まれていても低いカットオフで質の良いデータを持っている方が有利であるため、より良いバイオインフォマティクスは、この問題を軽減するために必要とされるであろう。
対立遺伝子頻度の検出および解釈は、腫瘍の割合の決意とゲノムの一部の領域の増幅バイアスの困難に困難な場合があります。ケース2において観察されるように加えて、50%を超える対立遺伝子頻度は、これは、ヘテロ接合性の消失(LOH)正常な対立遺伝子の喪失に起因する場合、いずれかの変異体読み出しの見かけの増加をもたらす、Aとして解釈され、検出され得ます変異対立遺伝子( 例えば 、2変異体および1通常のコピー)または他のメカニズムのゲイン。これらのメカニズムは、アレイ比較ゲノムハイブリダイゼーション(のaCGH 19)および/ またはSNP遺伝子型決定のアレイを利用することによって明らかにすることができる。20。
現在のターゲット濃縮方法は、非効率なハイブリッドキャプチャまたは単一のサンプルとターゲットシーケンシング読み取りオフ以上の複数の配列決定の報道のための必要性をもたらす多重PCR技術のいずれかの完全日の手続きに依存しています。近い将来に予想されるNGS分子腫瘍学のための追加のアプリケーションは、完全に自動化することが、非常に低い入力DNA( すなわち 、1未満NG)の量だけでなく、高度に分解した試料でサンプルを処理することができることができます簡単にライブラリー調製方法が含まれますDNA。これらの課題に対処するために、ほとんどのメソッドは、おそらく、PCRベースのいずれかになります多段階のPCR法や超並列シングルプレックスPCRアプローチです。また、個々のアンプリコンの分子バーコードは劇的背景シーケンシングノイズを低減し、より低い対立遺伝子頻度を達成するために、腫瘍細胞の低い割合を有するサンプルの検査を可能にし、circulatを取り込むに向かって移動することが示されています腫瘍細胞る。
癌標本における疾患関連変異の検出は、数十年の治療の標準となっています。歴史的には、遺伝子は、多くの場合、試験配列の末端に導く変異の同定と、一度に連続して、一つの遺伝子/エキソンを試験しました。 NGSの出現は、腫瘍形成に関連付けられている複数の変異の同定につながる並列に多くの癌に関連した複数の遺伝子の配列を決定するに偏りの少ないアプローチを可能にしました。癌における体細胞変異の検出のためのNGSの臨床的有用性がますます明らかです。実際、腫瘍サンプルのNGSベースの分析では、伝統的な、単一の遺伝子検査に挑戦する新しいパラダイムを表すが、臨床的有用性は非常にはっきりしています。臨床検査室は本日、この強力な技術を適用して慎重な方法の検証とテストの解釈を結婚するエキサイティングな機会を持っています。
Disclosures
The authors have nothing to disclose.
Acknowledgements
著者は、生産の原稿と支援の読み取りのためにダニエル・ワイルドの支援を感謝したいです。
Materials
| Genomic DNA ScreenTape | Agilent Technology | 5067-5365 | |
| Genomic DNA Reagents | Agilent Technology | 5067-5366 | |
| High Sensitivity D1000 ScreenTape | Agilent Technology | 5067-5584 | |
| High Sensitivity D1000 Reagents | Agilent Technology | 5067-5585 | |
| TapeStation 2200 | Agilent Technology | G2965A | |
| TapeStation Analysis Software | Agilent Technology | A.01.04 or higher | |
| 96-well Tube Storage Racks | Any Vendor | ||
| 15/50 ml Tube Rack | Any Vendor | ||
| 96-well Plate Rack | Any Vendor | ||
| Pipette, single-channel, 0.5–2.5 μL | Any Vendor | ||
| Pipette, single-channel, 1–10 μL | Any Vendor | ||
| Pipette, single-channel, 2–20 μL | Any Vendor | ||
| Pipette, single-channel, 10–100 μL | Any Vendor | ||
| Pipette, single-channel, 20–200 μL | Any Vendor | ||
| Pipette, single-channel, 100–1000 μL | Any Vendor | ||
| Serological Pipettor | Any Vendor | ||
| Vortexer | Any Vendor | ||
| Ice bucket | Any Vendor | ||
| Microcentrifuge (for tubes and strip tubes) | Any Vendor | ||
| Freezer, -20 °C | Any Vendor | ||
| 4 °C Refrigerator | Any Vendor | ||
| Water or Bead Bath | Any Vendor | ||
| Incubator (37 oC) | Any Vendor | ||
| Serological Pipettes, 1 mL | Any Vendor | ||
| Serological Pipettes, 5 mL | Any Vendor | ||
| Serological Pipettes, 10 mL | Any Vendor | ||
| Serological Pipettes, 25 mL | Any Vendor | ||
| Gloves | Any Vendor | ||
| Razor Blades/Scaples | Any Vendor | ||
| KimWipes | Any Vendor | ||
| 15 mL Conical Tube | Any Vendor | ||
| 50 mL Conical Tube | Any Vendor | ||
| Paper Towels | Any Vendor | ||
| 200 proof Ethanol | Any Vendor | Store in Flammable Cabinet | |
| 2-Propanol (Isopropanol) | Any Vendor | Store in Flammable Cabinet | |
| 25ml Reservoirs | Any Vendor | ||
| 10N NaOH | Any Vendor | ||
| Pipette, 8-channel, 1–10 μL | Any Vendor | ||
| Pipette, 8-channel, 10–100 μL | Any Vendor | ||
| Pipette, 8-channel, 20–300 μL | Any Vendor | ||
| Ice Bucket | Any Vendor | ||
| Water Squirt Bottle | Any Vendor | ||
| Alcohol Squirt Bottle | Any Vendor | ||
| Lens Cleaning Paper | Any Vendor | ||
| Plates, 96-well PCR, Semi-Skirted | Any Vendor | ||
| Tube strips, 8-well, 0.2 mL | Any Vendor | ||
| Agencourt AMPure XP Beads | Beckman Coulter | A63881 | |
| BioShake IQ or 3000-T elm | Bulldog Bio/Q.Instruments | 1808-0506/ 1808-0517 | |
| DropPlate96 S – LabChipDS | Caliper | 128876 | |
| DropPlate96 D – LabChipDS | Caliper | 132848 | |
| DropSense96 | Caliper (Trinean) | ||
| DropQuant Software | Caliper (Trinean) | ||
| Plate Sealing Film | Denville | B1212-5S | |
| Aluminum Seal Foil | Denville | B1212-6S | |
| Nuclease-Free, Pure Water System | EMD Millipore | ||
| 5424 centrifuge | Eppendorf | 22621408 | |
| 5804R centrifuge | Eppendorf | 22623508 | Both 15 ml tube and plate rotators, preferably a centrifuge that can go up to 2,500 x g. |
| Safe-Lock Tube 1.5 mL, Natural | Eppendorf | 22431021 | |
| 5 mL Tube, DNA LoBind Tube | Eppendorf | 30108310 | |
| 5430R Centrifuge | Eppendorf | 022620645 | Any plate rotator centrifuge will work |
| Hybex Microsample Incubator | Fisher Scientific | 1057-30-0 | |
| Hybex 0.2 mL Tube Block | Fisher Scientific | 1057-31-0 | |
| TruSeq Amplicon – Cancer Panel | Illumina | FC-130-1008 | 96 reactions |
| TruSeq Custom Amplicon | Illumina | PE-940-1011 | 96 reactions |
| TruSeq Custom Amplicon Index Kit | Illumina | FC-130-1003 | 96 Indices, 384 Samples |
| MiSeq Reagent Kit v3, 500 Cycles | Illumina | MS-102-3003 | |
| MiSeq Reagent Kit v2, 300 Cycles | Illumina | MS-102-2002 | |
| MiSeq Reagent Kit v2, 500 Cycles | Illumina | MS-102-2003 | |
| Experiment Manager | Illumina | 1.3 or higher | |
| MiSeq Reporter | Illumina | 2.0 or higher | |
| Sequencing Analysis Viewer | Illumina | 1.8 or higher | |
| TruSeq Index Plate Fixture and Collar Kit | Illumina | FC-130-1007 | |
| MiSeq v2 | Illumina | SY-410-1003 | |
| TruSeq Custom Amplicon Filter Plate | Illumina | FC-130-1006 | |
| Index Adapter Replacement Caps | Illumina | 11294657 | |
| Qubit 2.0 | Invitrogen | Q32866 | |
| Qubit 0.5 ml Tubes | Invitrogen | Q32856 | |
| Qubit dsDNA Broad Range Assay Kit | Invitrogen | Q32853 | |
| DynaMa6-96 Magnetic Stand, Side Skirted | Invitrogen | 120.27 | |
| GeneAmp PCR System 9700 (gold/silver block) | Life Technologies | N8050200 | |
| Gentra Puregene Blood Kit | Qiagen | 158489 | |
| Deparaffinization Solution (16ml) | Qiagen | 19093 | |
| Buffer ATL (4x50ml) | Qiagen | 939011 | |
| Protein Precipitation Solution (50 ml) | Qiagen | 158910 | |
| DNA Hydration Solution (100ml) | Qiagen | 158914 | |
| Glycogen Solution (500 μl) | Qiagen | 158930 | |
| Qiagen Proteinase K | Qiagen | 19133 | |
| Rnase (5ml) | Qiagen | 158924 | |
| Nuclease-Free Water (10 x 50 ml) | Qiagen | 129114 | |
| Pestles | USA Scientific | 1415-5390 | |
| TipOne RPT 10 ul elongated filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips). | USA Scientific | 1180-3810 | |
| TipOne RPT 100 ul natural, beveled filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1840 | |
| TipOne RPT 200 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-8810 | |
| TipOne RPT 20 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1810 | |
| TipOne RPT 1000 μl natural, graduated XL filter pipet tips in | USA Scientific | 1182-1830 |
References
- Gerlinger, M., et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 366 (10), 883-892 (2012).
- Campbell, P. J., et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 467 (7319), 1109-1113 (2010).
- Ding, L., et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 481 (7382), 506-509 (2012).
- Frampton, G. M., et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature Biotechnol. 31 (11), 1023-1031 (2013).
- Patel, J. P., et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 366 (12), 1079-1089 (2012).
- Forbes, S. A., et al. COSMIC (the Catalogue of Somatic Mutations in Cancer ): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 38 (Database Issue), 652-657 (2010).
- Shih, A. H., Abdel-wahab, O., Patel, J. P., Levine, R. L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 12 (9), 599-612 (2012).
- Liersch, R., Müller-Tidow, C., Berdel, W. E., Krug, U. Prognostic factors for acute myeloid leukaemia in adults – biological significance and clinical use. Br J Haematol. 165 (1), 17-38 (2014).
- Bacher, U., Schnittger, S., Haferlach, T. Molecular genetics in acute myeloid leukemia. Curr Opin Oncol. 22 (6), 646-655 (2010).
- Subramanian, J., Govindan, R. Lung cancer in "Never-smokers": a unique entity. Oncology (Williston Park). 24 (1), 29-35 (2010).
- Sakashita, S., Sakashita, M., Tsao, M. S. Genes and pathology of non-small cell lung carcinoma. Semin Oncol. 41 (1), 28-39 (2014).
- Arcila, M. E., Chaft, J. E., Nafa, K. Prevalence clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 18 (18), (2012).
- Gandhi, L., et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol. 32 (2), 68-75 (2014).
- Sheikhha, M. H., Awan, A., Tobal, K., Liu Yin, J. A. Prognostic significance of FLT3 ITD and D835 mutations in AML patients. Hematol J. 4 (1), 41-46 (2003).
- Mazières, J., et al. Lung cancer that harbors an HER2 mutation epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 31 (16), 1-8 (2014).
- Robinson, J. T., et al. Integrative Genomics Viewer. Nat Biotechnol. 29 (1), 495-500 (2011).
- Forbes, S. A., et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43 (Database issue), D805-D811 (2014).
- Daber, R., Sukhadia, S., Morrissette, J. J. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genetics. 206 (12), 441-448 (2013).
- Haraksingh, R. R., et al. Genome-Wide Mapping of Copy Number Variation in Humans: Comparative Analysis of High Resolution Array Platforms. PLoS ONE. 6 (11), e27859 (2011).
- de Leeuw, N., et al. SNP Array Analysis in Constitutional and Cancer Genome Diagnostics – Copy Number Variants, Genotyping and Quality Control. Cytogenet Genome Res. 135 (3-4), 212-221 (2011).
