While conventional bulk/real-time readouts can be used for both quantitative PCR and quantitative WGA assays (Figure 1), digital quantitative assays provide advantages (Table 1). In the method described, we read out digital assays in micro-droplet format with a simple bulk endpoint measurement (Figure 2). While this method is broadly applicable, we focus on quantitative WGA (MDA) because this method presents special challenges for conventional real-time assays.
To check whether our real-time PCR instrument could detect varying fractions of fluorescent microdroplets dispersed in oil, we measured the bulk fluorescence of synthetic mixtures of fluorescent and non-fluorescent droplets (Figure 2). The bulk fluorescence and positive droplet counts (independently assessed by fluorescence microscopy) scale linearly with the input fraction of fluorescent droplets as expected (Figure 3A). The experiment was performed three times, with independent droplet formation and mixing for each set.
To evaluate the theoretical quantification performance for “unknown” samples, we established linear standard curves using entirely positive and entirely negative control samples. With such droplet-lot-specific standard curves, we calculated the fraction of synthetic positive and negative droplet mixtures based on each sample’s bulk fluorescence. The results show good performance in droplet ratio quantification across two logs of dynamic range (R2 = 0.984; Figure 3B). The input analyte concentration is easily calculated from the fraction of positive or negative droplets38.
To test our method in a real quantitative WGA assay, we measured fluorescence levels and fraction of positive droplets of a digital droplet MDA assay with Lambda DNA across a wide range of concentrations (Figure 4). In Figure 4B, representative fluorescent images of the droplets are shown. Both bulk fluorescence and positive droplet fraction from the digital MDA samples scale as expected with the average template per droplet, indicating that the bulk readout can faithfully capture the result of a digital assay (R2 = 0.927). We repeated the experiment for each concentration, independent forming droplets and carrying out WGA for each replicate.
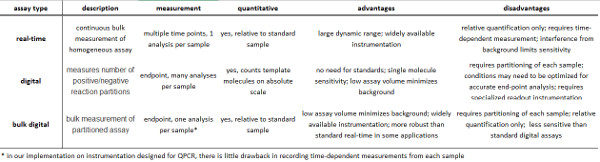
Table 1. Summary of real-time and digital assays. Please click here to view a larger version of this figure.
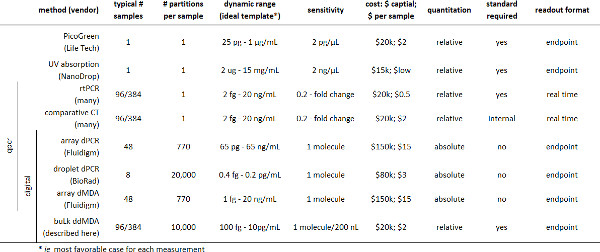
Table 2. Summary of existing methods for quantitating nucleic acids. Please click here to view a larger version of this figure.
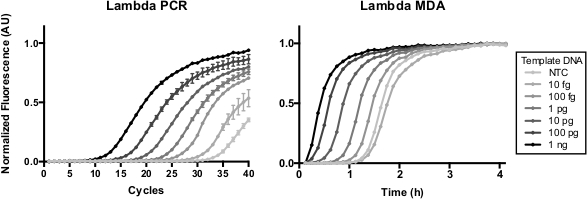
Figure 1. Real-time quantitative PCR and MDA of Lambda DNA. MDA is not discretized through temperature cycling like PCR, and is prone to preamplification if not prepared carefully on ice. Here, quantitative PCR and MDA were performed with increasing concentrations of Lambda DNA. Please click here to view a larger version of this figure.
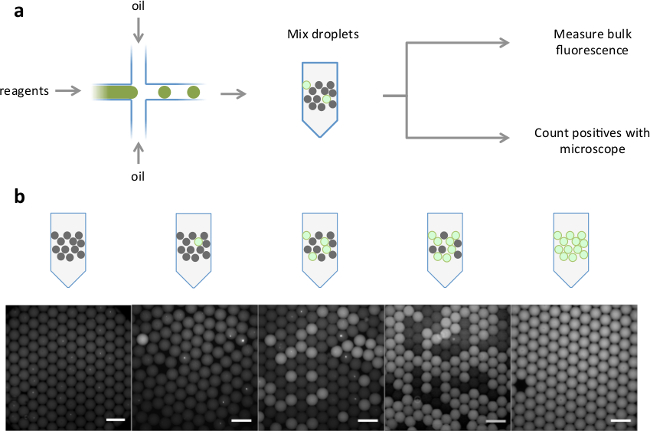
Figure 2. Schematic of analog readout of droplets and representative images. (A) Positive and negative droplets were generated from fluorescent and non-fluorescent reagents in a microfluidic device. The droplets were pre-mixed in different positive:negative ratios. Bulk fluorescence levels were measured with a standard real-time thermocycler, and fraction of positive droplets was determined by fluorescence microscopy. (B) Representative fluorescent overlaid images of increasing ratios of positive droplets (scale bar = 200 µm). Please click here to view a larger version of this figure.
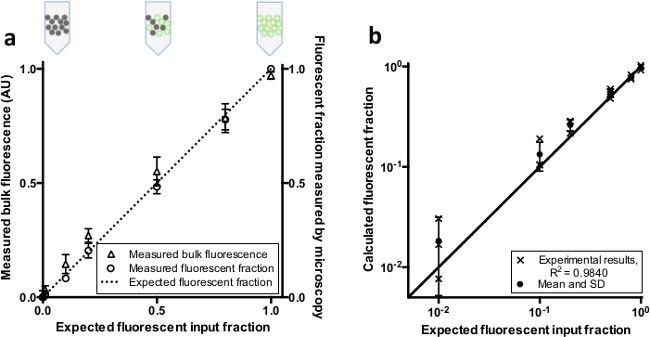
Figure 3. Bulk fluorescence and fluorescent fraction of combinations of separately prepared positive and negative droplets. (A) Fluorescent (positive) and non-fluorescent (negative) droplets were pre-mixed in different ratios. Comparison of the input fraction of positive droplets with the measured bulk fluorescence and measured positive droplet fraction (n = 3, bars represent +/- SD). The dotted line represents the expected fluorescent fraction given a linear relationship. (B) Comparison of predicted ratios based on standards to the expected fluorescent input fraction. The line indicates the expected value given a linear relationship. Please click here to view a larger version of this figure.
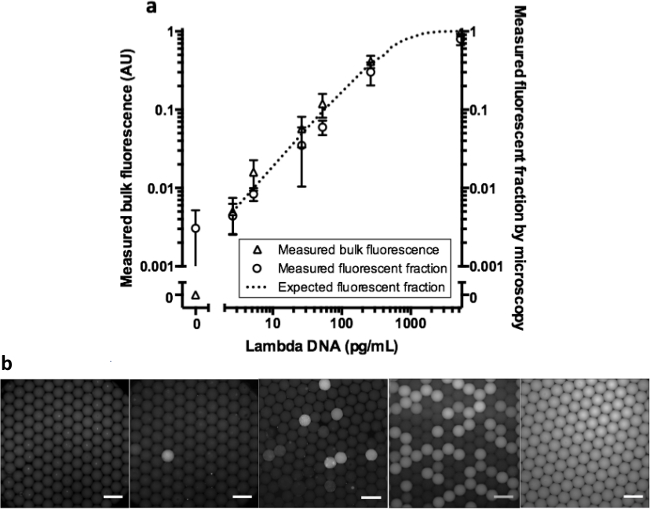
Figure 4. Bulk fluorescence and fluorescent fraction of a droplet digital MDA assay. Digital MDA was performed with increasing concentrations of template Lambda DNA (n = 3, error bars represent SD) and measured as described in Figure 2A. The line indicates the expected fluorescent fraction using the Poisson distribution to model the data from our experiment. b) Representative fluorescent overlaid images of droplets after reaction inactivation (scale bar = 200 µm) for increasing expected Lambda DNA molecules per droplet (NTC, 0.005, 0.05, 0.5, and 10). Please click here to view a larger version of this figure.
