Figure 1 and 2 show representative images of QD655 labeled, membrane-bound EGFR visualized in intact, fully hydrated COS-7 cells. The DIC image in Figure 1 a gives an impression of the membrane topography of the cells, and the corresponding fluorescence image in Figure 1b depicts the distribution of EGFR after 3 min of EGF-Biotin incubation. Figure 1A and B are each stitched together from two images recorded with a 40X air objective. The EGF activated EGFR is distributed over the entire cellular surface. In most cells a slight enhancement of fluorescence (Figure 1B) can be seen at the cell edges, indicating a locally increased occurrence of EGFR20. Control experiments conducted using a similar protocol verified specific EGFR labeling (data not shown). These controls included: 1) a control labeling without previous incubation of the cells with EGF-biotin, i.e., only incubation with STR-QDs, and 2) an incubation with non-biotinylated EGF and STR-QDs.
The three cells marked with rectangles were further investigated with ESEM-STEM. The ESEM-STEM images depicted in Figure 1C–E show low magnification overviews of these cells. These transmission images reflect whole cells in hydrated state with a thin layer of water residing over the cell placed on a silicon microchip with silicon nitride (SiN) viewing window. The vacuum chamber in the microscope contained water vapor, and the balance between sample temperature and pressure was adjusted to maintain a thin layer of liquid over the cells. The individual cells can be easily recognized on account of their shape and location on the SiN membrane window. The low-magnification ESEM-STEM images reveal fine structures, such as filopodia, extending from the cell edge towards neighboring cells, and some structures inside the thinner cell regions. Thick central cellular regions including the nucleus appear white because transmission through the sample is not possible for electrons of the used energy (30 keV).
High-resolution ESEM-STEM images were recorded in the thinner, peripheral regions. The spatial resolution for ESEM-STEM of nanoparticles in the thin regions of cells in a thin liquid layer was determined in a previous study6 to amount to 3 nm. Four high-resolution images shown in Figure 2A–D were recorded at the locations of the small rectangles in Figure 1C–E. The used magnification was sufficient to discern individual QDs, appearing as bright, bullet-shaped rods, each bound to an individual EGFR. The QD655 has typical dimensions of 6 x 14 nm2.
Figure 2A (corresponding to the rectangular area indicated on the cell in Figure 1C) shows QD labels on a membrane fold diagonally crossing the image. This membrane structure has a higher EGFR density than the surrounding membrane regions. At several locations, two labels were at close proximity. Two examples with distances of 20 and 24 nm are indicated with arrowheads. These pairs of labels are interpreted as belonging to EGFR dimers. Figure 2B gives an example from the edge of a cell (Figure 1D, left rectangle), EGFR monomers and dimers can be seen as well. Figure 2C (Figure 1C, right rectangle) was recorded of a region with a lower fluorescence signal, i.e., lower EGFR density. Nevertheless, EGFR was also found here in dimers as well. In addition, two clusters of 10 or 11 EGFRs were present (see ellipses). Figure 2D shows another example of a membrane fold (Figure 1E) with smaller clusters including 5-6 EGFRs, in addition to several monomers and dimers.
As an example of the kind of information that can be obtained from this data, the pair correlation function21 g(x) was determined for all label positions in Figure 2. Note that this analysis is not part of this protocol and the procedure was described elsewhere6,7. g(x) is a measure of the chance of a particle to be found within a certain radial distance x from a reference particle. g(x) = 1 represents a random distribution, and a larger value is evidence for clustering. The positions of a total of 210 labels were automatically detected in the four images of Figure 2A-D, and g(x) was calculated using a bin size of 5 nm and a smoothing filter with a bandwidth of 10 nm. The g(x) curve (Figure 3) shows a preferred center-to-center QD distance of 25 nm. EGFRs are thus not randomly oriented but a significant fraction of them resides at this preferred distance. From an approximate molecular model6 (Figure 3 inset) we estimate that the center-to-center distance between the QD and the EGF binding pocket amounts to ~14 nm, and center-to-center distance between the two QDs attached to a EGFR dimer to be ~27 nm (this value is likely to vary by a few nanometers due to the flexibility of the linker). The preferred label distance is thus consistent with the expected label distance for the EGFR dimer within the precision of the method. The g(x) curve is larger than unity for distance of up to 300 nm, indicating the presence of clusters, consistent with the data. This analysis shows that the stoichiometry of the EGFR can be studied with our method.
Figure 4 shows a similar result as Figure 2, except that the labeling was performed with EGF-QD800, and a 63X oil immersion objective was used for the fluorescence image. This data confirms that these typical results are also found when the EGFR labeling is done with QDs, which emit in the near red spectrum. Figure 4A is the fluorescence image of a representative cell, Figure 4B shows the same cell in ESEM-STEM overview mode and Figure 4C is a 150,000 x magnification image, recorded at the upper membrane border of the cell (see rectangle in Figure 4B). Similar to Figure 2A and D, the images capture QD-labeled EGFR on a membrane fold and shows monomeric, dimeric, and clustered receptors. Note that the electron dense QD core appears somewhat smaller and rounder than the cores of QDs emitting at 655 nm, consistent with their smaller core size22 of ~5 nm.

Figure 1. Correlative DIC, fluorescence and ESEM-STEM of EGF-QD655 labeled EGFR on fully hydrated COS-7 cells. (A) DIC image of the cells grown on the SiN membrane window area, giving a topographic impression of the plasma membrane. (B) Fluorescence image showing cells on the centrally located SiN membrane window (marked by the dashed rectangle). (C–E) Three ESEM-STEM low magnification images from the cells marked with rectangles in A and B. These cell overview images serve to determine the precise location of subsequently recorded high-resolution images. The location of four high-resolution images (shown in Figure 2A-D) are marked with white rectangles. Please click here to view a larger version of this figure.
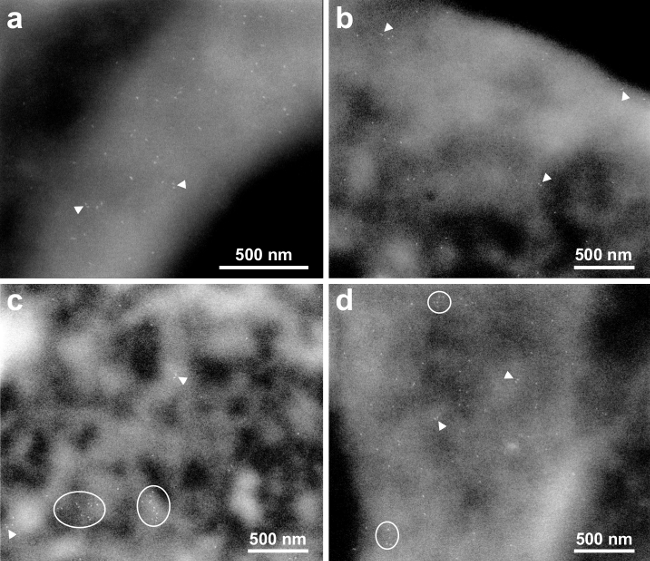
Figure 2. High-resolution ESEM-STEM images depicting membrane bound EGFRs. (A) Micrograph at 75,000X magnification of the area marked in Figure 1B. Many monomers are visible. Several dimers are indicated with the arrowheads. (B) and (C) images acquired at 50,000X magnification of the left, respectively right, membrane areas marked in Figure 1C. Clusters of EGFRs are outlined. (D) image recorded with 50,000X magnification at the location marked in Figure 1D. Please click here to view a larger version of this figure.
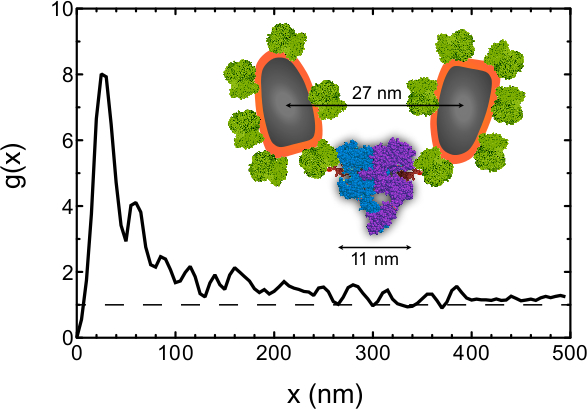
Figure 3. Pair correlation function g(x) as function of the radial distance x determined for the inter-particle distances of all 210 labels detected in Figure 2A-D. The peak at 25 nm indicates that a center-to-center QD distance has a much higher chance of occurring than random, whereby a g(x) = 1 indicates a random chance. The dashed line is a guide to the eye representing g(x) of a random distribution. The inset shows an approximate molecular model of the EGFR dimer with bound EGF and streptavidin coated QDs attached via biotin. The models of streptavidin, EGF and the EGFR were obtained from CPK models of the 1stp (streptavidin), 1EGF (EGF), 1NQL, 2JWA, 1M17, 1IVO and 2GS6 (EGFR) structures in the RCSB Protein Protein Databank, created by Jmol Version 12.2.15. The biotin model is as drawn in RCSB Ligand Explorer Version 1.0. Please click here to view a larger version of this figure.
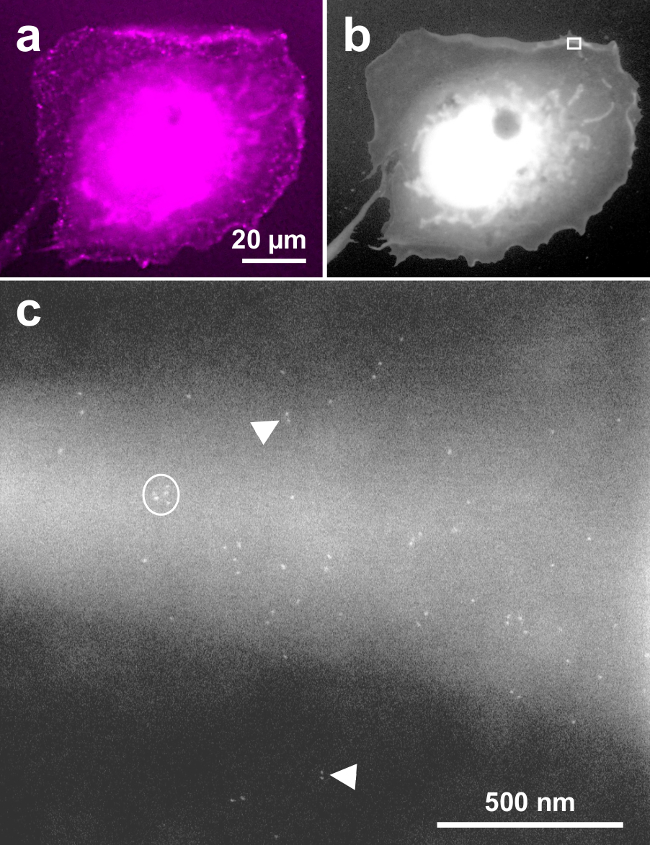
Figure 4. Exemplary COS-7 cell labeled with EGF-QD800 and imaged with correlative fluorescence and ESEM-STEM. (A) Fluorescence image, (B) low magnification overview ESEM-STEM image. (C) High-resolution image recorded at the location of the rectangle in b at 150,000X magnification. Monomeric, dimeric, and clustered EGFRs are detected similar to labeling with EGF-QD655. Please click here to view a larger version of this figure.
