A schematic diagram of the L1 retrotransposition vector is presented in Figure 1. The vector consists of a neomycin gene in antisense orientation to L1 ORFs that is interrupted by a globin intron in sense orientation and sandwiched by SD and SA sites. When stably integrated into a chromosome, L1 mRNA is transcribed from the combined CMV and L1-5'UTR promoter (Figure 1). During RNA processing, the globin intron is spliced out of the neomycin gene. The Neo-L1 RNA is packaged, translocated and integrated into the genome as either a full length or truncated insertion. As indicated, L1 retrotransposition can be tracked by FISH using probes specific for spliced neomycin (Figure 1).
Figure 2 shows a schematic representation of the neomycin probe, with the labeled probed that is larger in size and fluorescence deficient due to differences in excitation wavelengths. Note that the CY3 and FITC excite at different wavelengths than ethidium bromide stained DNA.
The density of metaphase chromosome spreads was determined by diluting the original stock solution into 1:2, 1:4, 1:8 and 1:16 dilutions and each spread evaluated for density, distribution and distance of spreads from the nucleus at 10X magnification (Figure 3A). The results show high density, clumpy and burst out spreads (Figure 3A-i-ii), as well as evenly distributed and well-spaced spreads (Figure 3A-iii-iv). Low density spreads with even distribution were chosen for subsequent analysis.
Chromosome spreads were stained with Hoechst dye and spread quality evaluated based on the length of chromosomes, roundness of the spreads and inter-chromosome distance (Figure 3B). These indices were influenced by the length of time the cells were treated with colcemid, hypotonic solution, Carnoy Fixative solution and/or the manner in which the cells were busted to release chromosomes. If cells were incubated for a short period in hypotonic solution, chromosome spreads became tightly knotted and individual chromosomes were difficult to visualize (Figure 3B-i). On the other hand, longer incubation in hypotonic solution resulted in rupture of the nuclei, scattering of chromosomes, and/or loss of chromosomes (Figure 3B-ii). Longer incubations in colcemid increased the number of cells in metaphase, but lead to condensation of chromosomes (Figure 3B-iii). As such, a good quality chromosome spread requires optimal incubation periods in both colcemid and hypotonic KCl solution. In our hands, a 90 min incubation in colcemid, 20 in hypotonic solution at 37 °C and the use of a hot steam to burst the cells were found to be optimal conditions for generation of high quality spreads (Figure 3B-iv).
Chromosome spreads were stained for L1 retrotransposition using probes that target SNeo. The probe was labeled with both FITC and CY3 to show that in both cases L1 retrotransposition is seen only in cells expressing wild type L1 (Figure 4). No staining was seen in cells expressing the vector backbone alone (Figure 4; compare top and bottom panels for each fluorophore). In our hands, probe signal was detected in 80-90% of nuclei, with the overwhelming majority of the signal representing single retrotransposition events. We only scored retrotransposition in nuclei with more than one insertion and spread quality was always examined before FISH.
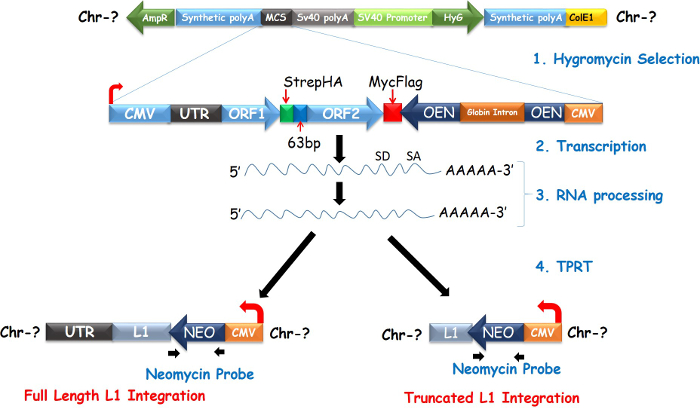
Figure 1. Schematic diagram of L1 retrotransposition vector. Diagram depicts the process of target prime reverse transcription leading to full or truncated L1 insertions. Please click here to view a larger version of this figure.
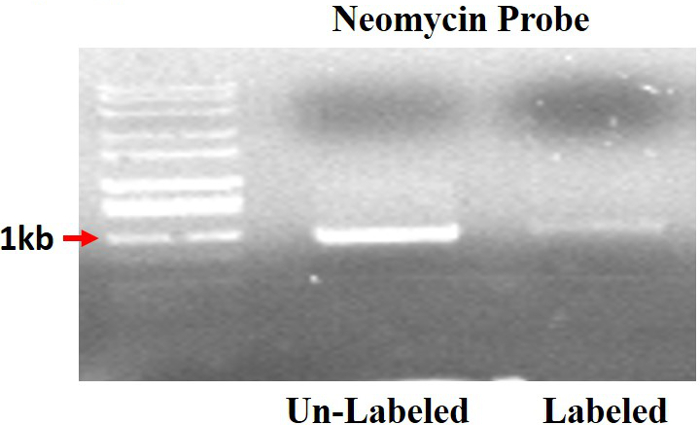
Figure 2. Labeled SNeo and unlabeled neomycin probes. The labeled probe exhibits less fluorescence and is larger in size compared to the unlabeled probe. Please click here to view a larger version of this figure.
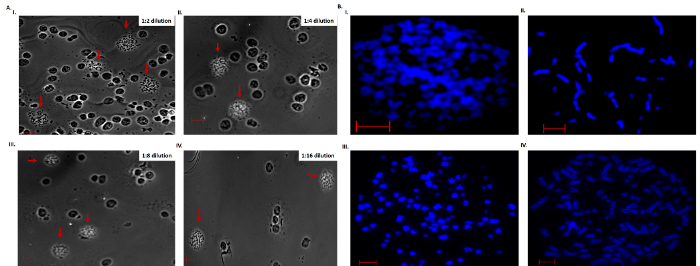
Figure 3. Phase-contrast (Bright Field) and Hoechst stain chromosome spreads. A) Shows dilution of spreads to obtain well-spaced preparations. Scale bar is 50 µm. B) Hoechst stain chromosome showing the influence of colcemid, hypotonic solution, Carnoy Fixative solution and the bursting on spread quality. Scale bar is 10 µm. Please click here to view a larger version of this figure.
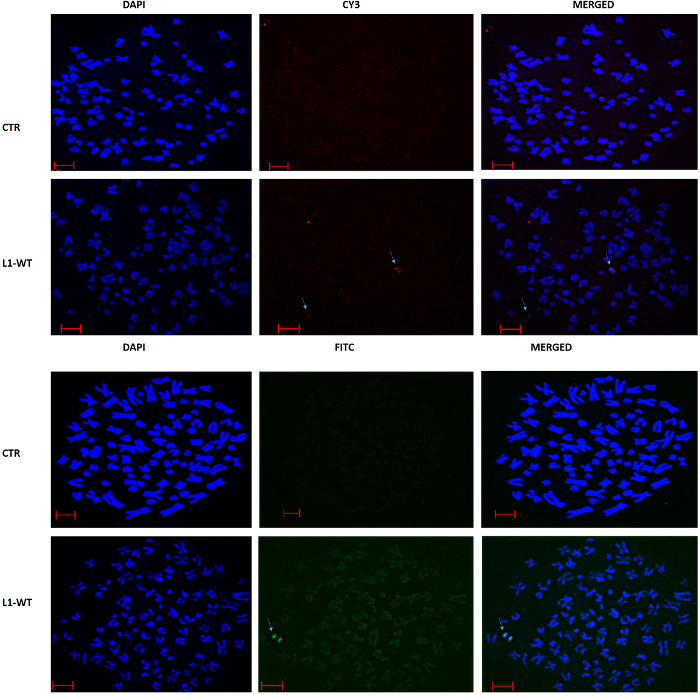
Figure 4. FISH analysis of L1 retrotransposition. Staining of SNeo is absent in control cells, but present in cells expressing L1 wildtype vector. Scale bar is 10 µm. Please click here to view a larger version of this figure.
