The following figures illustrate representative results from the ChIP-exo protocol presented here. In contrast to traditional ChIP-seq methodologies with few enzymatic steps, ChIP-exo requires eleven sequentially dependent enzymatic reactions (Figure 1). Thus, care must be taken at each step to ensure that each reaction component is added to its respective reaction master mix. We recommend generating a formulaic spreadsheet based on the reaction Tables to automatically perform the reaction master mix calculations, printing the resulting tables, and then checking off each item after it is added to the master mix.
We employ a number of quality control measures throughout the protocol to ensure high quality sequencing results (Figure 2). Each ChIP reaction contains three basic components: 1) sonicated chromatin extract from cells of interest, 2) an antibody directed against the protein of interest, and 3) ProteinG (or ProteinA) resin to immobilize the precipitated immune complexes. Obtaining high quality sonicated chromatin extracts (Figure 2A) can be quite challenging since sonication conditions must be optimized for each cell type and sonication instrument. It is worth spending time to optimize this step because the highest quality libraries start with a sonication result that yields DNA fragments between 100 – 500 bp (Figure 2A, "+" lane). Since formaldehyde crosslinks are labile and formaldehyde itself has a limited shelf-life, we use an electrophoretic mobility shift assay (Figure 2A, "-" lane) to verify that the sonicated extracts contain intact protein-DNA crosslinks, evident as super-shift of the DNA fragments. To assess background signal we routinely perform a "mock" ChIP that omits antibody from the ChIP reaction. A high quality ChIP-exo library preparation will have very little, if any, background signal in the "mock" ChIP ("-") relative to the specific ChIP antibody, in this case directed against Pol II. As seen in Figure 2B, traditional ChIP-seq libraries have substantially more background signal than ChIP-exo. Lastly, prior to sequencing, ChIP-exo library quality is assessed on the bioanalyzer (Figure 2C – D). Analysis accurately measures the library size distribution and detects contaminating adapter dimers (denoted by arrow) that run at 125 bp. If adapter dimers are present, they will reduce the sequencing bandwidth. Therefore, we recommend an additional bead cleanup that will efficiently remove DNA fragments less than 200 bp.
ChIP-exo is a powerful functional genomic technique because it is the only method capable of spatially resolving divergent, initiating, paused, and elongating RNA polymerase II on a genome-wide scale (Figure 3) 11. Since these adjacent binding events are tens of base pairs apart, ChIP-seq is unable to distinguish these binding events with resolving power of several hundred base pairs.
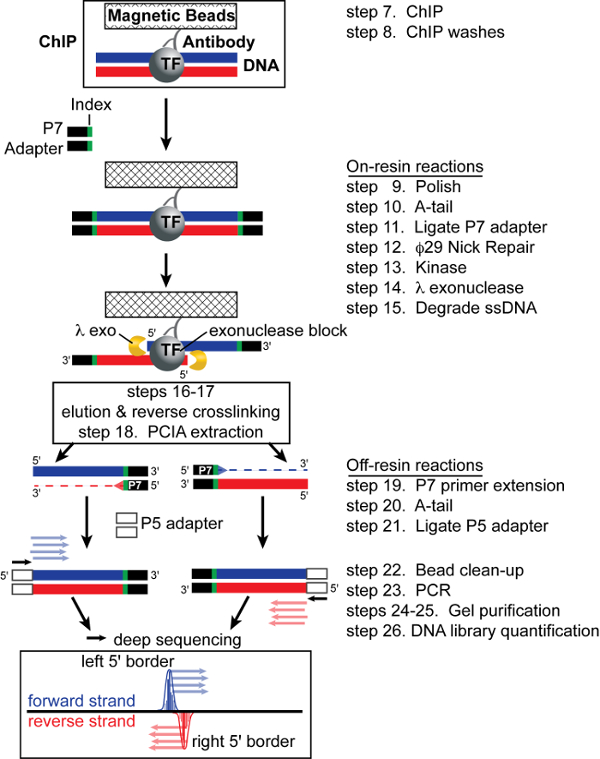
Figure 1: ChIP-exo Schematic. After ChIP, the P7 adapter is ligated to the sonication borders. Lambda exonuclease then trims DNA 5' to 3' to the crosslink point, thereby footprinting the protein-DNA interaction. After elution and crosslink reversal, primer extension synthesizes duplex DNA. Lastly, ligation of the P5 adapter marks the left and right exonuclease borders and the resulting library is subjected to high-throughput sequencing. Mapping the 5' ends of the sequence tags to the reference genome demarcates the exonuclease barrier and thus the precise site of protein-DNA crosslinking. Figure modified from Rhee and Pugh 17. Please click here to view a larger version of this figure.
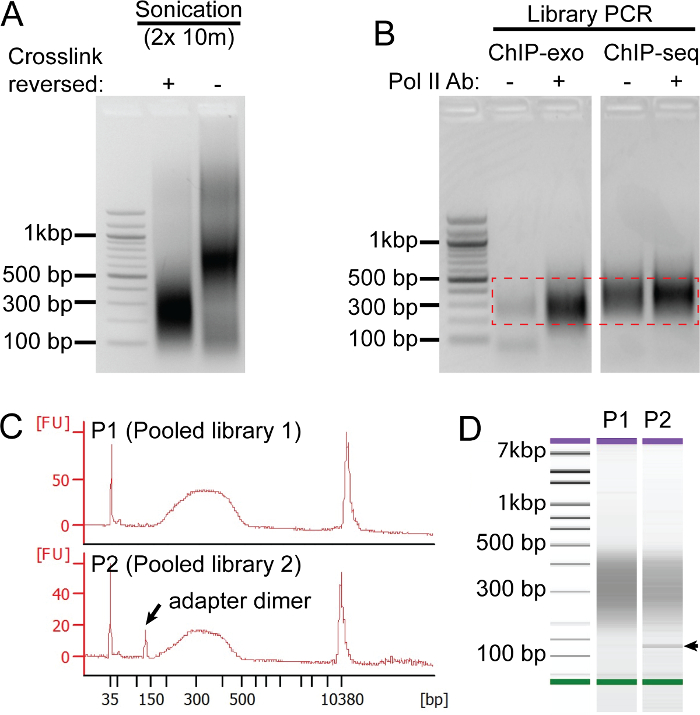
Figure 2: ChIP-exo Quality Controls. Panels A-C show representative results from unrelated experiments. (A) The quality of the sonicated chromatin extract (from the human HCC1806 breast cancer cell line) is assessed by agarose gel electrophoresis on extracts with (+) and without (-) the crosslinks reversed. Intact crosslinks will cause protein-DNA complexes to migrate slower. Thus, running extract without crosslinks reversed (-) allows for the quality of the formaldehyde crosslinks to be assessed, which are critical for a successful ChIP. (B) Comparison of a mock IP (-) and Pol II (+) ChIP-exo and ChIP-seq library preparations after 21 cycles of PCR amplification. After PCR, DNA fragments 200-500 bp are excised (denoted by red hashed box) and purified using a gel extraction kit. (C-D) In panel C, the top trace represents an ideal trace from a pooled library (P1), and the bottom trace shows a pooled library (P2) that contains adapter dimers. Panel D shows the DNA density plot corresponding to the P1-2 library traces from panel C (arrow denotes adapter dimer band). Please click here to view a larger version of this figure.
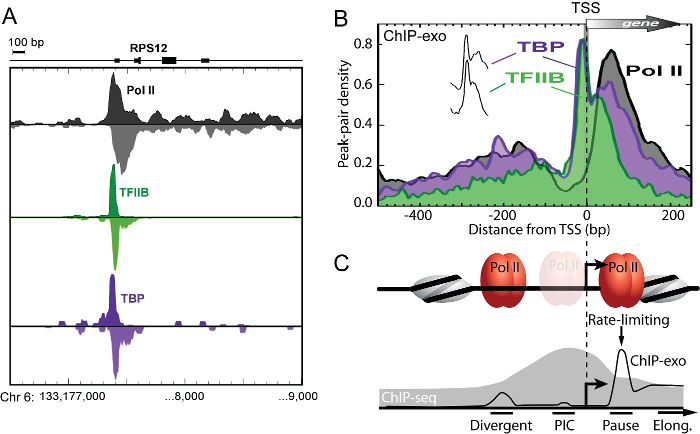
Figure 3: ChIP-exo Spatially Resolves Distinct Bidirectional Transcription Initiation Complexes. (A) Smoothed distribution of strand-separated ChIP-exo tag 5' ends for Pol II, TFIIB, and TBP at the human RPS12 gene in proliferating K562 cells. (B) Averaged ChIP-exo patterns around the closest RefSeq TSS. Peak-pair tags were aligned to the TSS gene-by-gene, binned in non-overlapping 10bp intervals relative to the TSS, and then the average peak-pair density value across all TFIIB-occupied (n = 6,511) genes was plotted as a percent of the total. The "spikes" of TBP and TFIIB are indiscernible (vertically offset in inset). (C) Model based on panel B data, illustrating distinct transcription initiation complexes resolved by ChIP-exo (black trace). Pol II occupied two separate resolvable locations that coincided with sites of divergent transcription initiation ("Divergent") and "Pause" sites. This clear spatial separation of Pol II complexes indicates that divergent transcripts arise from distinct initiation complexes. The vast majority Pol II crosslinked about 50 bp downstream of the TSS at the "Pause" site, where it is expected to pause after initiating transcription. Pol II was most depleted 20 – 60 bp upstream of the TSS where the pre-initiation complex ("PIC") forms, indicating that on average it likely spends less time there than at the paused sites. This suggests that in most (but not necessarily all) cases, once Pol II is recruited, it rapidly clears the promoter and assumes a paused-state approximately 30 – 50 bp downstream of the TSS, consistent with the observation that Pol II pause release is a rate-limiting step in transcription. These adjacent initiation complexes are unresolvable by ChIP-seq (illustrated by gray fill trace) since its resolution is limited to a few hundred base pairs. Figure modified from Pugh and Venters 11. Please click here to view a larger version of this figure.
Supplemental File 1: Additional Information. Please click here to download this file.
| Reagent | Volume (ml) | [Final] |
| 1 M HEPES-KOH (pH 7.5) | 50 | 50 mM |
| 5 M NaCl | 28 | 140 mM |
| 0.5 M EDTA | 2 | 1 mM |
| 100% Glycerol | 100 | 10% |
| 10% NP40 | 50 | 0.50% |
| 10% Triton X100 | 25 | 0.25% |
| ddH2O | Fill to 1 L |
Table 1. Recipe for Lysis Buffer 1. Filter using 0.22 μm filter. Store in 50 ml tubes at 4 ˚C. Add 100 μl CPI stock to 50 ml of buffer just prior to use.
| Reagent | Volume (ml) | [Final] |
| 1 M Tris-HCl (pH 8) | 10 | 10 mM |
| 5 M NaCl | 40 | 200 mM |
| 0.5 M EDTA | 2 | 1 mM |
| 0.5 M EGTA | 1 | 0.5 mM |
| ddH2O | Fill to 1 L |
Table 2. Recipe for Lysis Buffer 2. Filter using 0.22 μm filter. Store in 50 ml tubes at 4 ˚C. Add 100 μl CPI stock to 50 ml of buffer just prior to use.
| Reagent | Volume (ml) | [Final] |
| 1 M Tris-HCl (pH 8) | 10 | 10 mM |
| 5 M NaCl | 20 | 100 mM |
| 0.5 M EDTA | 2 | 1 mM |
| 0.5 M EGTA | 1 | 0.5 mM |
| 10% Deoxycholate | 10 | 0.10% |
| N-lauroylsarcosine | 5 g | 0.5% (w/v) |
| ddH2O | Fill to 1 L |
Table 3. Recipe for Lysis Buffer 3. Filter using 0.22 μm filter. Store in 50 ml tubes at 4 ˚C. Add 100 μl CPI stock to 50 ml of buffer just prior to use.
| Reagent | Volume (ml) | [Final] |
| 10x PBS | 50 | 1x |
| Bovine Serum Albumin | 2.5 g | 0.50% |
| ddH2O | Fill to 500 |
Table 4. Recipe for Blocking Buffer. Filter using 0.22 μm filter. Store in 50 ml tubes at 4 ˚C. Add 100 μl CPI stock to 50 ml of buffer just prior to use.
| Reagent | Volume (ml) | [Final] |
| 1 M HEPES (pH 7.5) | 25 | 50 mM |
| 0.5 M EDTA (pH 8) | 1 | 1 mM |
| 10% Sodium Deoxycholate | 35 | 0.70% |
| 10% NP40 | 50 | 1% |
| 1 M LiCl | 250 | 500 mM |
| ddH2O | Fill to 500 |
Table 5. Recipe for RIPA Buffer. Filter using 0.22 μm filter. Store in 50 ml tubes at 4 ˚C. Add 100 µl CPI stock to 50 ml of buffer just prior to use.
| Reagent | Volume (ml) | [Final] |
| 1 M Tris-Cl (pH 7.5) | 2.5 | 50 mM |
| 0.5 M EDTA | 1 | 10 mM |
| 20% SDS | 2.5 | 1% |
| ddH2O | Fill to 50 |
Table 6. Recipe for ChIP Elution Buffer. Filter using 0.22 μm filter. Store at RT.
| Reagent | Volume (ml) | [Final] |
| 1 M Tris-Cl (pH 7.5) | 0.5 | 10 mM |
| ddH2O | Fill to 50 |
Table 7. Recipe for TE Buffer. Filter using 0.22 μm filter. Store at 4 ˚C.
| Volume (μl) | [Final] | |
| 100 μM ExA2-iX | 75 | 15 μM |
| 100 μM ExA2-33 | 75 | 15 μM |
| 1 M Tris (pH 7.5) | 50 | 100 mM |
| 5 M NaCl | 5 | 50 mM |
| ddH2O | 295 | – |
| Total volume | 500 |
Table 8. P7 Adapter Annealing mix.
| Volume (μl) | [Final] | |
| 100 μM ExA1-58 | 75 | 15 μM |
| 100 μM ExA1-13 | 75 | 15 μM |
| 1 M Tris (pH 7.5) | 50 | 100 mM |
| 5 M NaCl | 5 | 50 mM |
| ddH2O | 295 | – |
| Total volume | 500 |
Table 9. P5 Adapter Annealing mix.
| Temp (˚C) | Time |
| 95 | 5 min |
| 72 | 5 min |
| 65 to 60 ramp decline | 5 min |
| 55 to 50 ramp decline | 3 min |
| 45 to 40 ramp decline | 3 min |
| 30 | 3 min |
| 20 | 3 min |
| 10 | 3 min |
| 4 | Forever |
Table 10. Adapter Annealing Program.
| 1x (μl) | [Final] | |
| ddH2O | 39.8 | |
| 10x Reaction Buffer 2 | 5 | 1x |
| 100 µM ATP | 0.5 | 1 mM |
| 3 mM dNTPs | 1.7 | 100 μM |
| 3 U/μl T4 polymerase | 1 | 3 U |
| 5 U/μl Klenow | 1 | 5 U |
| 10 U/μl T4 Polynucleotide Kinase | 1 | 10 U |
| Total reaction volume | 50 |
Table 11. Polishing master mix.
| 1x (μl) | [Final] | |
| ddH2O | 42.3 | |
| 10x Reaction Buffer 2 | 5 | 1x |
| 3 mM dATP | 1.7 | 100 μM |
| 5 U/μl Klenow 3'-5' exo minus | 1 | 5 U |
| Total reaction volume | 50 |
Table 12. A-tailing master mix.
| 1x (μl) | [Final] | |
| ddH2O | 41 | |
| 100 mM ATP | 0.5 | 1 mM |
| 10x Reaction Buffer 2 | 5 | 1x |
| 400 U/μl T4 DNA Ligase | 1.5 | 600 U |
| Reaction mix volume | 48 | |
| 15 mM Index Adapter | 2 | 30 picomoles |
| Total reaction volume | 50 |
Table 13. P7 Adapter Ligation master mix.
| 1x (μl) | [Final] | |
| ddH2O | 41 | |
| 10x Φ-29 Buffer | 5 | 1x |
| 3 mM dNTPs | 2.5 | 150 μM |
| 10 U/μl Φ-29 polymerase | 1.5 | 15 U |
| Total reaction volume | 50 |
Table 14. Φ-29 Nick Repair master mix.
| 1x (μl) | [Final] | |
| ddH2O | 43.5 | |
| 100 mM ATP | 0.5 | 1 mM |
| 10x Reaction Buffer 2 | 5 | 1x |
| 10 U/μl T4 Polynucleotide Kinase | 1 | 10 U |
| Total reaction volume | 50 |
Table 15. Kinase Reaction master mix.
| 1x (μl) | [Final] | |
| ddH2O | 43 | |
| 10x Lambda Buffer | 5 | 1x |
| 5 U/μl Lambda exonuclease | 2 | 10 U |
| Total reaction volume | 50 |
Table 16. Lambda Exonuclease Reaction master mix.
| 1x (μl) | [Final] | |
| ddH2O | 44 | |
| 10x Reaction Buffer 2 | 5 | 1x |
| 30 U/μl RecJf exonuclease | 1 | 30 U |
| Total reaction volume | 50 |
Table 17. RecJf Nuclease Reaction master mix.
| 1x (μl) | [Final] | |
| ddH2O | 6.45 | |
| 10x Φ-29 Buffer | 2 | 1x |
| 3 mM dNTP | 1.3 | 200 μM |
| 20 μM P7 Primer | 0.25 | 0.25 μM |
| Reaction mix volume | 10 | |
| EtOH precipitated sample | 10 | |
| Total reaction volume | 20 |
Table 18. P7 Primer Extension Reaction master mix.
| Temp (˚C) | Time |
| 95 | 5 min |
| 65 | 5 min |
| 30 | 2 min |
| 30 | Hold until Φ-29 is added |
| 30 | 20 min |
| 65 | 10 min |
| 4 | Forever |
Table 19. P7 Primer Extension program.
| 1x (μl) | [Final] | |
| ddH2O | 5 | |
| 10x Reaction Buffer 2 | 3 | 1x |
| 3 mM dATP | 1 | 0.1 mM |
| 5 U/μl Klenow 3’ to 5’ exo minus | 1 | 5 U |
| Reaction mix volume | 10 | |
| Primer extended sample | 20 | |
| Total reaction volume | 30 |
Table 20. A-tailing master mix.
| 1x (μl) | [Final] | |
| ddH2O | 11.5 | |
| 10x T4 Ligase Buffer | 5 | 1x |
| 15 μM ExA1-58/13 adapter | 2 | 30 picomoles |
| 400 U/μl T4 DNA Ligase | 1.5 | 600 U |
| Reaction mix volume | 20 | |
| A-tailed sample | 30 | |
| Total reaction volume | 50 |
Table 21. P5 Adapter Ligation master mix.
| 1x (μl) | [Final] | |
| 5x PCR Buffer | 10 | 1x (2 mM MgCl2) |
| 10 mM of each dNTP | 1 | 200 μM each |
| 20 μM P1.3 Primer | 1.25 | 0.5 μM |
| 20 μM P2.1 Primer | 1.25 | 0.5 μM |
| 2 U/μl Hot Start polymerase | 0.5 | 1 U |
| Reaction mix volume | 14 | |
| Bead eluted sample | 36 | |
| Total reaction volume | 50 |
Table 22. PCR.
| Temp (˚C) | Time | Cycles |
| 98 | 30 sec | 1 |
| 98 | 10 sec | 15-21 (depending on ChIP efficiency) |
| 52 | 30 sec | |
| 72 | 20 sec | |
| 72 | 2 min | 1 |
| 4 | Forever | Hold |
Table 23. PCR program.
| Per DNA Standard (μl) | Per Sample (μl) | |
| 1:200 diluted buffer/dye mix | 190 | 198 |
| DNA standards | 10 | |
| ChIP-exo library | 2 | |
| Total volume | 200 | 200 |
Table 24. Quantification.
