नोट: प्रोटोकॉल कहीं भी ठहर सकता है ।
1. मूल सेटअप एक बिस्तर, चोटियों प्रारूप, या BigWig 7 फ़ाइल के जीनोम में इनपुट होने के लिए डेटा युक्त । फ़ाइल का एक्सटेंशन नाम होना चाहिए & #34; bed & #34;, & #34; broadpeaks & #34; & #34; narrowpeaks & #34;, या & #34; bigWig & #34; क्रमश. & #8203; नोट: इन प्रकार की फ़ाइलों के ज़िप्ड संस्करण भी काम करेंगे । genemo.org जाने के लिए एक इंटरनेट ब्राउज़र का उपयोग करें । सबसे आम इंटरनेट ब्राउज़र चलाने में सक्षम किसी भी ऑपरेटिंग सिस्टम GeNemo. का उपयोग करने में सक्षम होना चाहिए ड्रॉपडाउन मेनू का उपयोग कर के खिलाफ खोज करने के लिए जो प्रजातियों चुनें । वर्तमान में उपलब्ध प्रजातियों में मानव और माउस शामिल हैं । अपलोड उपयोगकर्ता एक यूआरएल या एक प्रत्यक्ष अपलोड का उपयोग कर फ़ाइल । BigWig फ़ाइलें केवल url अपलोड विधि के साथ काम करते हैं । बिस्तर और चोटियों प्रारूप फ़ाइलें दोनों विधियों के साथ काम करते हैं (अब के रूप में मुख्य डेटा के रूप में चढ़ाव फ़ाइलें अपलोड नहीं किया जा सकता).
2. वैकल्पिक सेटअप खोज किया जाता है जब ई-मेल द्वारा खोज परिणाम प्राप्त करने के लिए संबंधित बॉक्स में एक ईमेल पता प्रदान करें । & #8203; नोट: जब जीनोम का एक बड़ा हिस्सा खोज और/या पटरियों की एक बड़ी संख्या के खिलाफ (नीचे देखें), यह अनुशंसित है कि उपयोगकर्ता उसके ईमेल प्रदान करता है, क्योंकि खोज एक लंबा समय लग सकता है । उदाहरण के लिए, एक १०० megabase खोज 15 एस के आसपास लेता है । खोज के पूर्ण होने पर प्रदान किए गए ई-मेल पते पर एक लिंक भेजा जाएगा । लिंक एक खोज के पूरा होने के बाद 7 दिनों में समाप्त हो जाएगा । प्रदान एक bigwig फ़ाइल या चढ़ाव प्रदर्शन फ़ाइल किसी url से हो सकता है । यह प्रदर्शन फ़ाइल परिणामों को प्रभावित नहीं करेगी; यह केवल परिणामों के साथ दिखाया जाएगा. संबंधित बॉक्स में (गुणसूत्र और आधार जोड़ी पदों सहित) एक खोज रेंज निर्दिष्ट करें । गुणसूत्र की सूची, आधार युग्म, और अंत आधार जोड़ी शुरू करते हैं । उपयोग & #39; chrN & #39; गुणसूत्र प्रारूप के लिए, जहां & #39; N & #39; गुणसूत्र संख्या/पत्र (1, 2, & #8230; X, या Y) है । आधार जोड़े के लिए, बस संख्या में टाइप करें । सभी तीन प्रविष्टियों के बीच रिक्त स्थान शामिल हैं, या एक बृहदांत्र (गुणसूत्र संख्या और पहली आधार जोड़ी के बीच:), और/ उदाहरणार्थ: chr1:1000000-2000000, chr1 १०००००० २००००००, chr1 1000000-2000000, chr1:1000000 २००००००. नोट: चरण २.१-२.३ वैकल्पिक है.
3. डाटा चयन 4. खोज और परिणाम डेटा चयन के बाद & #34; Search & #34; बटन क्लिक करें । खोज में कुछ समय लग सकता है । खोज पूरी होने के बाद, उपयोगकर्ताओं को परिणाम पृष्ठ में विभिंन बक्से दिखाई देंगे । प्रत्येक बॉक्स जीनोम के एक खंड का प्रतिनिधित्व करता है, जहां उपयोगकर्ता & #39; s डेटा फ़ाइल में एक या अधिक ट्रैक के साथ एक निकट मिलान प्रतिमान है उपयोगकर्ता ने क्वेरी की है । यदि कोई बॉक्स दृश्यमान नहीं हैं, तो अधिक प्रकार के ट्रैक खोजने या खोज श्रेणी को समान इनपुट फ़ाइल से बड़ा करने का प्रयास करें । सब कुछ के बिना यह करने के लिए एक आसान तरीका क्लिक कर रहा है & #34; & #9776; & #34; बटन लोगो के बगल में । यह एक साइडबार है कि उपयोगकर्ता खोज को संशोधित करने की अनुमति देता है खुल जाएगा । परिणाम पृष्ठ के तल पर & #34;D ownload bed file & #34; बटन पर क्लिक करके एक बेड फाइल के रूप में निर्यात किया जा सकता है । परिणामों को विज़ुअलाइज़ करने के लिए प्रत्येक बॉक्स के ऊपर दाईं ओर विज़ुअलाइज़ करें बटन पर क्लिक करे. दाईं तरफ विज़ुअलाइज़ेशन पैनल में, एकाधिक चीज़ें डेटा सहित प्रदर्शित की जाती हैं, जो उपयोगकर्ता इनपुट फ़ाइल को शामिल करती है, प्रदर्शन फ़ाइल अगर एक से बाहर किया गया था, पटरियों मिलान, और कुछ डिफ़ॉल्ट पटरियों । परिणाम से, उपयोगकर्ता ज्ञात एन्कोडेड dataset के विरुद्ध आगे की जाँच के लिए दिए गए डेटासेट की तुलना कर सकते हैं । उपयोगकर्ता क्वेरी परिणामों के संदर्भ को देखने के लिए UCSC जीन का उल्लेख भी कर सकते हैं । यदि एकाधिक सेल लाइनों/ऊतकों से पटरियों का चयन किया जाता है, तो उपयोगकर्ता दिए गए डेटासेट और एनकोडिंग डेटासेट के बीच समानता की ऊतक विशिष्टता के बारे में अंतर्दृष्टि प्राप्त करने के लिए ऐसे परिणामों का उपयोग कर सकते हैं. परिणाम पृष्ठ पर , उपयोगकर्ता किसी भी पटरियों पर खींचें के लिए ऊपर या जीनोम के बहाव को स्थानांतरित कर सकते हैं; जब माउस कर्सर निर्देशांक पर है, तो उपयोगकर्ता माउस व्हील का उपयोग करें और/या में और बाहर ज़ूम कर सकते हैं । 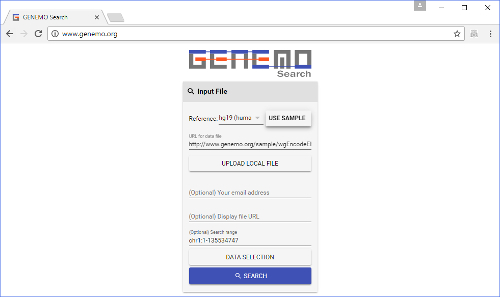 चित्रा 1 : GeNemo & #39; एस के आवश्यक क्षेत्रों के साथ सामने पृष्ठ । & #160; एक उपयोगकर्ता को प्रजातियों, खोज फ़ाइल और खोज रेंज इनपुट की जरूरत है, और पटरियों का चयन करें वह के खिलाफ खोज करने के लिए चाहता है । ईमेल पता और प्रदर्शन फ़ाइल वैकल्पिक हैं । & #160; इस फिगर का बड़ा वर्जन देखने के लिए यहां क्लिक करें ।
चित्रा 1 : GeNemo & #39; एस के आवश्यक क्षेत्रों के साथ सामने पृष्ठ । & #160; एक उपयोगकर्ता को प्रजातियों, खोज फ़ाइल और खोज रेंज इनपुट की जरूरत है, और पटरियों का चयन करें वह के खिलाफ खोज करने के लिए चाहता है । ईमेल पता और प्रदर्शन फ़ाइल वैकल्पिक हैं । & #160; इस फिगर का बड़ा वर्जन देखने के लिए यहां क्लिक करें ।  चित्रा 2 : ट्रैक चयन खिड़की. & #160; यह सामने पृष्ठ पर & #34;D ata चयन & #34; बटन पर क्लिक करके लाया जाता है । यहाँ, उपयोगकर्ता इनपुट फ़ाइल के विरुद्ध खोज करने के लिए ट्रैक का चयन करें । कुछ ट्रैक्स पहले से डिफॉल्ट रूप से चयनित हैं । & #160; इस फिगर का बड़ा वर्जन देखने के लिए यहां क्लिक करें । डेटा चयन बटन क्लिक करने के बाद, ( यानी , क्वेरी में जोड़ने के लिए) के विरुद्ध खोज करने के लिए ट्रैक के किस प्रकार का चयन करें । ट्रैक संग्रह दुनिया भर में प्रयोगशालाओं से कई विभिन्न डेटासेट शामिल हैं. पटरियों की सूची के रूप में काफी लंबा है, उपयोगकर्ताओं को ट्रैक चयन की सुविधा के लिए फ़िल्टर बटन (शीर्ष पर) का उपयोग करना चाहते हो सकता है । पटरियों प्रयोग, ऊतक, सेल लाइन और/ द्वारा फ़िल्टर किया जा सकता है ट्रैक चयन निष्पादित करने में मदद करने के लिए नीचे दिए गए पांच बटन हैं: सभी का चयन करें, कोई नहीं का चयन करें, जोड़ें, फ़िल्टर, बाहर । का चयन सभी & #34; र & #34; चय क कोई भी & #34; य है । & #34; Add & #34; बटन वर्तमान में चयनित ट्रैक्स को क्वेरी में जोड़ता है । यह तर्क गेट & #34 के रूप में कार्य करता है; या & #34;. ध्यान दें कि उपरोक्त फ़िल्टर ( उदा. , कुछ प्रयोग, ऊतक, सेल लाइन या लैब्स) का चयन करने से खोज क्वेरी में स्वचालित रूप से संगत ट्रैक नहीं जुड़ते हैं । उपयोगकर्ताओं को पहले पटरियों ( जैसे , मस्तिष्क, जिगर के नीचे ऊतक) का चयन करना होगा, और फिर उन्हें क्वेरी में जोड़ने के लिए & #34; Add & #34; बटन पर क्लिक करें. ट्रैक का चयन करते समय, नोट करें कि केवल फ़िल्टर विंडो में खोले गए टैब में निर्दिष्ट फ़िल्टर्स खोज क्वेरी पर लागू किए जाएंगे. अंय टैब्स पर चयनों को फ़िल्टर विंडो में सहेजा जाएगा, लेकिन खोज क्वेरी पर लागू नहीं होगा । & #34; फ़िल्टर & #34; बटन केवल वर्तमान में क्वेरी में फ़िल्टर विंडो में चयनित ट्रैक के प्रकार बनाए रखता है और अन्य सभी प्रकार की ट्रैक्स को निकालता है. यह तर्क गेट & #34 के रूप में कार्य करता है; और & #34;. मूलतः, & #34; फिल् & #34; पटरियों की दो श्रेणियों ( जैसे , कुछ प्रयोगशालाओं के साथ कुछ ऊतकों) के बीच बातचीत के चयन की अनुमति देता है. ध्यान दें कि & #34; फ़िल्टर & #34; यदि वे पहले से ही क्वेरी में नहीं हैं, तो चयनित प्रकार के ट्रैक्स को क्वेरी में न जोड़ें. & #34; छोडी & #34; बटन सभी प्रकार की ट्रैक्स निकालता है जो वर्तमान में क्वेरी से फ़िल्टर विंडो में चयनित हैं । यह तर्क gate & #34 के रूप में कार्य करता है; न & #34;, विरोध में & #34; फ़िल्टर & #34; फ़ंक्शन. नु, त & #34; छोडी & #34; कोई भी ट्रैक्स वर्तमान में फ़िल्टर विंडो में क्वेरी करने के लिए चयनित नहीं जोड़ नहीं है । चित्रा 3 : फ़िल्टर विंडो . इस पर क्लिक करके लाया जाता है & #34; फ़िल्टर & #34; बटन ट्रैक चयन विंडो पर । यहाँ, उपयोगकर्ताओं को एक ही समय में कई पटरियों का चयन कर सकते हैं, रिश्तेदार आसानी के साथ. & #160; इस फिगर का बड़ा वर्जन देखने के लिए यहां क्लिक करें । चित्रा ४ : फ़िल्टर फ़ंक्शन का उपयोग कैसे करें. & #160; इस फिगर का बड़ा वर्जन देखने के लिए कृपया यहां क्लिक करें । क्वेरी के लिए इच्छित ट्रैक जोड़ने के बाद, नीचे दाईं ओर & #34; Update & #34; बटन क्लिक करें । यह डेटा का चयन करने के लिए दो तरीके को समायोजित करने के लिए आवश्यक है: व्यक्तिगत डेटा पटरियों का चयन या फ़िल्टरिंग/ द & #34; रिसेट दृ & #34; बटन मानव/माउस भ्रूण स्टेम कोशिकाओं में जीन अभिव्यक्ति विनियमन से संबंधित डिफ़ॉल्ट ट्रैक करने के लिए क्वेरी रीसेट करता है । नोट: ट्रैक का चयन via & #34 के खिलाफ खोजा जा;D ata चयन & #34; वैकल्पिक है लेकिन अनुशंसितकारण डिफ़ॉल्ट खोज ट्रैक सबसे अधिक संभावना उपयोगकर्ता & #39; s आवश्यकताओं के लिए अनुकूल नहीं हैं । चित्रा ५ : परिणाम पृष्ठ . इस विशेष खोज ३६३ मिलान क्षेत्रों लौट आए । प्रथम मिलान क्षेत्र को प्रदर्शित करके, प्रत्येक परिणामी क्षेत्र बॉक्स के नीचे बाईं ओर & #34; SHOW & #34; बटन क्लिक करके किया जा सकता है । प्रदर्शन विंडो के बाएं भाग पर यह देखा जा सकता है कि दो डेटा फ़ाइलें (इनपुट और चयनित ट्रैक) संकेत शक्ति पैटर्न में समान हैं । & #160; कृपया click यहां इस आंकड़े का एक बड़ा संस्करण देखने के लिए ।
चित्रा 2 : ट्रैक चयन खिड़की. & #160; यह सामने पृष्ठ पर & #34;D ata चयन & #34; बटन पर क्लिक करके लाया जाता है । यहाँ, उपयोगकर्ता इनपुट फ़ाइल के विरुद्ध खोज करने के लिए ट्रैक का चयन करें । कुछ ट्रैक्स पहले से डिफॉल्ट रूप से चयनित हैं । & #160; इस फिगर का बड़ा वर्जन देखने के लिए यहां क्लिक करें । डेटा चयन बटन क्लिक करने के बाद, ( यानी , क्वेरी में जोड़ने के लिए) के विरुद्ध खोज करने के लिए ट्रैक के किस प्रकार का चयन करें । ट्रैक संग्रह दुनिया भर में प्रयोगशालाओं से कई विभिन्न डेटासेट शामिल हैं. पटरियों की सूची के रूप में काफी लंबा है, उपयोगकर्ताओं को ट्रैक चयन की सुविधा के लिए फ़िल्टर बटन (शीर्ष पर) का उपयोग करना चाहते हो सकता है । पटरियों प्रयोग, ऊतक, सेल लाइन और/ द्वारा फ़िल्टर किया जा सकता है ट्रैक चयन निष्पादित करने में मदद करने के लिए नीचे दिए गए पांच बटन हैं: सभी का चयन करें, कोई नहीं का चयन करें, जोड़ें, फ़िल्टर, बाहर । का चयन सभी & #34; र & #34; चय क कोई भी & #34; य है । & #34; Add & #34; बटन वर्तमान में चयनित ट्रैक्स को क्वेरी में जोड़ता है । यह तर्क गेट & #34 के रूप में कार्य करता है; या & #34;. ध्यान दें कि उपरोक्त फ़िल्टर ( उदा. , कुछ प्रयोग, ऊतक, सेल लाइन या लैब्स) का चयन करने से खोज क्वेरी में स्वचालित रूप से संगत ट्रैक नहीं जुड़ते हैं । उपयोगकर्ताओं को पहले पटरियों ( जैसे , मस्तिष्क, जिगर के नीचे ऊतक) का चयन करना होगा, और फिर उन्हें क्वेरी में जोड़ने के लिए & #34; Add & #34; बटन पर क्लिक करें. ट्रैक का चयन करते समय, नोट करें कि केवल फ़िल्टर विंडो में खोले गए टैब में निर्दिष्ट फ़िल्टर्स खोज क्वेरी पर लागू किए जाएंगे. अंय टैब्स पर चयनों को फ़िल्टर विंडो में सहेजा जाएगा, लेकिन खोज क्वेरी पर लागू नहीं होगा । & #34; फ़िल्टर & #34; बटन केवल वर्तमान में क्वेरी में फ़िल्टर विंडो में चयनित ट्रैक के प्रकार बनाए रखता है और अन्य सभी प्रकार की ट्रैक्स को निकालता है. यह तर्क गेट & #34 के रूप में कार्य करता है; और & #34;. मूलतः, & #34; फिल् & #34; पटरियों की दो श्रेणियों ( जैसे , कुछ प्रयोगशालाओं के साथ कुछ ऊतकों) के बीच बातचीत के चयन की अनुमति देता है. ध्यान दें कि & #34; फ़िल्टर & #34; यदि वे पहले से ही क्वेरी में नहीं हैं, तो चयनित प्रकार के ट्रैक्स को क्वेरी में न जोड़ें. & #34; छोडी & #34; बटन सभी प्रकार की ट्रैक्स निकालता है जो वर्तमान में क्वेरी से फ़िल्टर विंडो में चयनित हैं । यह तर्क gate & #34 के रूप में कार्य करता है; न & #34;, विरोध में & #34; फ़िल्टर & #34; फ़ंक्शन. नु, त & #34; छोडी & #34; कोई भी ट्रैक्स वर्तमान में फ़िल्टर विंडो में क्वेरी करने के लिए चयनित नहीं जोड़ नहीं है । चित्रा 3 : फ़िल्टर विंडो . इस पर क्लिक करके लाया जाता है & #34; फ़िल्टर & #34; बटन ट्रैक चयन विंडो पर । यहाँ, उपयोगकर्ताओं को एक ही समय में कई पटरियों का चयन कर सकते हैं, रिश्तेदार आसानी के साथ. & #160; इस फिगर का बड़ा वर्जन देखने के लिए यहां क्लिक करें । चित्रा ४ : फ़िल्टर फ़ंक्शन का उपयोग कैसे करें. & #160; इस फिगर का बड़ा वर्जन देखने के लिए कृपया यहां क्लिक करें । क्वेरी के लिए इच्छित ट्रैक जोड़ने के बाद, नीचे दाईं ओर & #34; Update & #34; बटन क्लिक करें । यह डेटा का चयन करने के लिए दो तरीके को समायोजित करने के लिए आवश्यक है: व्यक्तिगत डेटा पटरियों का चयन या फ़िल्टरिंग/ द & #34; रिसेट दृ & #34; बटन मानव/माउस भ्रूण स्टेम कोशिकाओं में जीन अभिव्यक्ति विनियमन से संबंधित डिफ़ॉल्ट ट्रैक करने के लिए क्वेरी रीसेट करता है । नोट: ट्रैक का चयन via & #34 के खिलाफ खोजा जा;D ata चयन & #34; वैकल्पिक है लेकिन अनुशंसितकारण डिफ़ॉल्ट खोज ट्रैक सबसे अधिक संभावना उपयोगकर्ता & #39; s आवश्यकताओं के लिए अनुकूल नहीं हैं । चित्रा ५ : परिणाम पृष्ठ . इस विशेष खोज ३६३ मिलान क्षेत्रों लौट आए । प्रथम मिलान क्षेत्र को प्रदर्शित करके, प्रत्येक परिणामी क्षेत्र बॉक्स के नीचे बाईं ओर & #34; SHOW & #34; बटन क्लिक करके किया जा सकता है । प्रदर्शन विंडो के बाएं भाग पर यह देखा जा सकता है कि दो डेटा फ़ाइलें (इनपुट और चयनित ट्रैक) संकेत शक्ति पैटर्न में समान हैं । & #160; कृपया click यहां इस आंकड़े का एक बड़ा संस्करण देखने के लिए ।
