1. Device Fabrication
- Download the attached mask file (Supplemental File 1) and generate a chrome mask using a commercial service or in-house facility. As the smallest dimension on the device is 10 µm (actuator membrane thickness), ensure that the mask has sufficiently high resolution, within ± 0.25 µm, to reliably produce the features.
- Follow standard SU-8 photolithography methods (e.g., references24,25,26) to fabricate the mold for subsequent production of PDMS devices; a summary of the steps is listed below.
NOTE: SU-8 is a photosensitive material, which crosslinks upon exposure to ultraviolet light (maximum absorption ~ 365 nm). Use the manufacturer instructions as a starting point to determine the processing parameters for the soft-bake, exposure (UV), and post-bake.- Deposit SU-8 2002 onto a silicon wafer and spin-coat it to an approximate thickness of 2 µm, for better adhesion of small structures. Soft-bake on a hot plate for 1 min at 95 °C, slightly over-expose (UV) the entire surface (~ 100 mJ/cm2), and post-bake on a hot plate for 2 min at 95 °C.
- Spin-coat a 47-µm thick layer of SU-8 2050 for defining the device features. Use a spin speed of 500 rpm for 15 s (130 rpm/s acceleration) and then 2,000 rpm for 90 s (260 rpm/s acceleration). If necessary, repeat and adjust the spin speed to get the proper thickness of photoresist.
- Soft-bake on a hot plate for 2 min at 65 °C, and then for 7 min at 95 °C. Expose the photoresist according to the manufacturer instructions (~ 160 mJ/cm2) using a chrome mask and long-pass filter to ensure straight side walls.
- Remove the wafer and post-bake on a hot plate for 1 min at 65 °C, and for 7 min at 95 °C.
- Dissolve unexposed SU-8 with developer, and clean with 2-propanol.
- Bake the wafer on a hotplate at 180 °C for 30 min (hard-bake) to stabilize photoresist features.
- Make a PDMS replica from the SU-8 model using standard replica molding methods27.
- Treat the SU-8 mold with trichloromethylsilane (TCMS) vapor to reduce PDMS adhesion (silanization).
CAUTION: TCMS is toxic and water-reactive.- Place the patterned wafer in a wafer rack within a bell-jar vacuum desiccator in a fume hood free of water or water-soluble reagents.
- Under the hood, use a dropper to apply 1 drop of TCMS to a glass dish and place inside the desiccator.
- Close the desiccator lid and allow TCMS vapor to coat the wafer for at least 20 min.
- Vent and then open the desiccator. Open the bell jar lid and remove the wafer using plastic tweezers. Place into a Petri dish or aluminum foil boat for PDMS replica molding. Dispose of TCMS-coated materials in the proper hazardous waste.
- Mix PDMS (ratio of 10:1) using 40 g of the base polymer and 4 g PDMS curing agent.
- Pour the mixture over the SU-8 mold and degas the mixture for at least 30 min in a vacuum chamber.
- Cure for at least 6 h at 70 °C in an oven.
- Treat the SU-8 mold with trichloromethylsilane (TCMS) vapor to reduce PDMS adhesion (silanization).
- Lift PDMS off the wafer by setting the wafer horizontal on a flat surface and carefully peeling the PDMS off. Do not bend the wafer upwards during this step, as it can break the mold.
NOTE: Incomplete adhesion of SU-8 to the wafer or incomplete silanization can result in destruction of the SU-8 structures during this step. - Cut PDMS around the devices into individual chips such that the devices will fit on a 24 mm x 60 mm coverslip.
- Using a 1-mm biopsy punch, make holes in the inlet, two outlets, and six actuators using a 1-mm biopsy punch. Aim for the 2 mm-diameter circles around the edges of the device.
- Bond PDMS chips to glass cover slips.
- Expose both surfaces to 80 W oxygen plasma (30 s).
- Gently place the exposed PDMS surface onto the exposed surface of the cover slip for a conformal seal.
- Anneal the device on a hot plate (100 °C, 10 min).
NOTE: Insufficient bonding can lead to the failure of the device due to leakage.
2. Preparation of the Microscope
NOTE: Transgenic animals: express a calcium indicator such as GCaMP6s28 or other genetically encoded activity probe in the neuron(s) of interest (e.g., TRN); co-express an activity-independent fluorescent protein emitting at a different wavelength to correct for small lateral and out-of-focus movement artifacts that arise due to mechanical stimulation. The automated analysis software tracks and compensates for movement-induced changes in the intensity of the activity probe. The worm strains GN69223 (or AQ323629) express GCaMP6s (or GCaMP6m) and the calcium-independent tagRFP under the control of mec-7 promotor. Additionally, GN692 contains the mutation lite-1(ce314), which prevents activation of TRNs due to the blue light sensor lite-130 during excitation of the GCaMP6s fluorescence23.
- Set up a microscope system for simultaneous excitation of GCaMP and RFP.
- Use either a continuous light source and a dual band excitation filter that transmits only cyan and yellow light, or use a light source that emits only wavelengths in a defined bandwidth such as simultaneous cyan (0.77 mW) and yellow (1.21 mW) LED excitation sources for simultaneous excitation of the calcium-dependent and independent fluorescence, respectively.
- Use a digital camera to enable recording of microscopy images.
NOTE: Most cameras come with a software for image acquisition. Alternatively, there are freely available software that can be used to control the camera and potentially also other parts of the microscopy system. - Adjust excitation intensity based on fluorescence intensity to avoid camera saturation.
- Use a fluorescence cube that depending of the choice of the excitation source needs to be equipped with an excitation filter.
NOTE: A 488-nm GFP and 580-nm mCherry excitation filter was used here.- Add a beam splitter to the cube for reflecting cyan and yellow light and transmitting green and red light.
- Equip the microscope with a 10X objective and a high-magnification objective (e.g., 63X/1.32 NA oil) to focus the excitation light onto the sample.
- Mount a beam splitter in front of the camera for simultaneous recording of the GCaMP and the calcium-independent signal. Ensure that the beam splitter has a dichroic mirror (long pass, cut-off at 570 nm) to separate green and red light using one emission filter for green (passband centered at 525 nm with 50 nm width) and one emission filter for red light (passband centered at 632 nm with 60 nm width).
- Project green and red fluorescence onto the upper half (green) and lower half (red), respectively of the camera chip (see Figure 3). This orientation is a prerequisite for the provided analysis software.
3. Animal Preparation
- Prepare age-synchronized young adult or adult day one C. elegans, as described previously by Porta-de-la-Riva et al.31
- Prepare the microfluidic chip.
- Connect the gravity flow reservoir (~ 60 cm above chip level) containing filtered (0.2-µm polyethersulfone syringe filter) M9 buffer to one outlet of the chip. Connect the other outlet to one outlet of a two-outlet waste container, i.e., a filter flask. Connect the other outlet of the waste container to a peristaltic pump.
NOTE: Use polyethylene (PE) tubing for all connections and use metal tube fittings to connect the PE tubing to the chip. This way all waste solutions will be flushed out of the chip and collected in the waste container. The flow through the outlet channels creates a gentle suction that will keep the worm snug during the later trapping process. - Prepare interconnects consisting of 50-mm long PE tubing (0.9652-mm OD, 0.5842-mm ID) with metal tubing (gauge size 23TW, 0.635-mm OD) on both ends. Press-fit these interconnects into each of the six actuation fingers and into the worm inlet (see Figure 1). Leave these interconnectors in the chip, as repeated removal leads to wear on the PDMS holes.
- Connect the gravity flow reservoir (~ 60 cm above chip level) containing filtered (0.2-µm polyethersulfone syringe filter) M9 buffer to one outlet of the chip. Connect the other outlet to one outlet of a two-outlet waste container, i.e., a filter flask. Connect the other outlet of the waste container to a peristaltic pump.
- Place the chip on the microscope. Pick 2–5 worms into a drop of filtered (0.2-µm syringe filter) M9 buffer and use a 1-mL syringe to draw them up into a PE tubing (0.9652-mm OD, 0.5842-mm ID) connected to a 1-mL syringe by pulling the plunger gently. Keep the animals in the PE tube and not in the syringe.
NOTE: Too many worms or unfiltered solutions in the chip can lead to clogging. - Connect the PE tubing of the syringe to the interconnect at the worm inlet (Figure 1 and Figure 2) of the chip. Activate the gravity flow by opening the valve and start the peristaltic pump. Then gently press the syringe plunger of the syringe to move the animals into the trapping channel while observing the channel under a microscope with a 10X lens in brightfield mode.
NOTE: Occasionally-appearing air bubbles do not pose a problem; they are usually automatically moved into the outlet. Several animals can be 'parked' in the waiting chamber and used sequentially.- After loading the animal into the trapping channel (see also Figure 2), adjust its position by gently pushing or pulling the plunger of the syringe to position the head of the animal in the tapered shape of the front of the channel.
- Make sure the worm (adult day 1) has the right size to be trapped in the chip.
- If the worm does not fill the entire cross-section of the channel from the nose to near the end of the body (not including the tip of the tail) or the worm moves along the axis of the channel without moving the plunger of the syringe, remove the worm by pressing the plunger of the syringe until it disappears from the trapping channel and load a new one (see step 3.8).
- Switch to fluorescence mode of the microscope and a higher magnification lens (40X or higher) and check whether the neurite of the neuron of interest comes to lie across the diaphragm of one of the actuators. If not, adjust the position of the animal by pulling or pushing the plunger. If that does not help, remove the worm and load a new one.
- Focus on the cell body of the neuron of interest and connect the interconnect of the actuator closest to the neuron on the anterior side of the cell body to a programmable pressure pump using PE tubing.
- If the experiment requires measuring the distance between the actuator and the neuron, move the field of view so both are in the field of view and the channelwall is parallel to the upper and lower edge of the image.
- Define a pressure protocol using the programmable pressure pump.
NOTE: This protocol can be adjusted to the desired experiment.- Start with a constant pressure of 0 kPa for a time corresponding to at least 50 images of the image sequence (necessary for normalization). For an imaging rate of 10 Hz this corresponds to 5 s. Add a desired stimulus waveform and pressure and define the length of the stimulus as a second step.
NOTE: For stimulating the TRNs, a sinusoidal waveform of (i.e., 75 kPa, 10 Hz) superimposed with a step (i.e., 275 kPa) is recommended as it generates a large neuronal response. - If the experiment requires additional stimuli, include a period of at least 10 s at a constant pressure of 0 kPa in between stimuli.Before switching the pressure pump on, make sure the instrument pressure is at constant 0 kPa to avoid accidental stimulation of the worm before the actual experiment.
- Start with a constant pressure of 0 kPa for a time corresponding to at least 50 images of the image sequence (necessary for normalization). For an imaging rate of 10 Hz this corresponds to 5 s. Add a desired stimulus waveform and pressure and define the length of the stimulus as a second step.
- Run the imaging and pressure protocol.
- In the image acquisition software of the camera, set up an image sequence at 10 Hz using an exposure time of 100 ms for the duration of the pressure protocol. Adjust the excitation intensity such that the maximal fluorescence increase does not saturate the camera. Save the images as a *.tif file with at least 50 images before the first stimulus for normalization.
- Start the imaging protocol in the image acquisition software and the pressure protocol in the software of the programmable pump. During recording, observe whether the neuron of interest is the brightest spot in an area of 10 x 10 pixels, does not move farther than 10 pixels in sequential images, and stays in the field of view during the recording.
NOTE: If this is not done, the analysis software will fail. Bright spots (i.e., autofluorescence) around the neuron make clean recordings of the neurons difficult. To correct this, remove the worm from the trap and load a new worm into the chip. If the worm moves too much, try a new recording of the same worm and observe the worm motion. If the problem persists the worm might be too small to be trapped. In this case remove this worm and load a slightly bigger worm into the trap. - If desired, record signals from multiple neurons in the field of view simultaneously as long as the neurons are separated by at least 10 pixels during the entire recording.
- To investigate habituation, perform the experiment repetitively on an animal.
- Remove the worm from the trapping channel.
- If it is desired to keep the worm for subsequent studies, disconnect the outlets of the chip towards the gravity flow and the waste container. Press the plunger of the syringe gently until the entire worm is pushed through the trapping channel into the flow channel.
- Continue pressing the syringe plunger until the animal appears in a droplet outside the chip. Disconnect the syringe including the PE tubing from the worm inlet, use it to aspirate the worms in the droplet and transfer it onto an agar plate.
- If it is not desired to keep the worm, remove the worm by pressing the plunger of the syringe until the entire worm is pushed through the trapping channel into the flow channel; the gravity flow and the suction of the peristaltic pump will carry the animal out of the chip and into the waste container.
4. Analysis
- Download and install the newest Fiji version32.
- Download and install the newest java SDK version.
NOTE: If necessary, delete or rename the java folder inside the Fiji folder for Fiji to use the newly installed java compiler. - Open Fiji software.
- Check whether the software is running the newly installed java compiler by opening 'Plugins| Utilities| ImageJ| Properties'.
- Download the pokinganalyzer*.java file (https://github.com/HFehlauer/Poking-Analyzer, see Supplementary File 2).
- Drag and drop the .java file into the software window to open it as a plugin.
- Compile and run the pokinganalyzer*.java file by pressing Ctrl+R keys in Fiji; a graphical user interface (Poking Analyzer) will open (Figure 3).
- Click on the "Help" tab and read about the requirements of the software and how to perform the analysis. To begin, select the "Open video" tab. Specify the video file location for the analyzer: click the "Open a Video" button, navigate to the location of the video, and open it.
NOTE: The software starts with this tab opened. If it is not open when starting a new analysis click on the "Open video" tab. If the video is in a.tif format, the program will internally open the video and display the first image in the lower part of the interface. If the image is not of the video that needs to be analyzed, click on the "Open a video" button to open another video. - To continue, click on the "Define ROIs" tab. In this tab, define the position of the TRN. Note that the first image of the opened video is displayed in the lower part of this tab. Click on the "Define TRN" button, and click on the neuron in the upper part of the displayed image which should display the GCaMP fluorescence.
- Optional: If the actuator is visible in the upper half of the image the program can automatically track the distance between the neuron and the actuator if desired. For this, check the "Define the actuator?"-box, click on the "Define Actuator" button, and click on the actuator in the upper half of the image.
- Click on the "Analysis" tab. Define the image acquisition rate. Either enter a number in the field or click the up or down buttons to increase or decrease the number. If in the previous tab the "Define the actuator?"-box is checked, define the camera cell size, the magnification, and the binning factor, so the program can calculate the distance.
- Click on the "Start Analysis" button.
NOTE: The program will now track the neuron by its fluorescence in the image sequence in the upper half (calcium dependent) and in the lower half (calcium independent) of the image sequence. To account for neuron motion in the plane of recording the program will investigate an area of 10 x 10 pixels around the location of the neuron in the previous image to find the position of the neuron in the following image. It will calculate the fluorescence in both halves by correcting for the background fluorescence (Fbg) and dividing by the fluorescence in the first 50 images (F0):
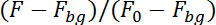
Fluorescence changes in the green, activity-dependent channel (FCa2+) that are caused by neuron motion out of the plane of recoding are then corrected using the fluorescence in the red, activity-independent channel (Fcorr) and the pre-stimulus standard deviation of the calcium-dependent (sdCa2+) and independent fluorescence (sdcorr) by:
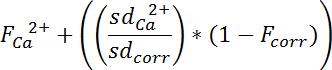
- The program will show its progress in the status bar in the lower part of the interface; when the status bar reaches 100%, click on the "Results" tab. If the status bar stops before 100%, the program will give an error message indicating the reason the program could not analyze the video.
- If the signal to noise ratio is too low, the program cannot identify the neuron; adjust the imaging parameters in subsequent recordings.
- If the neuron leaves the field of view during the recording, the program cannot track it anymore and will stop. For subsequent recordings, place the neuron in the middle of the field of view at the beginning of the recording.
- If some regions in the field of view are oversaturated, the automated analysis will produce invalid results. For subsequent recordings, make sure the fluorescence signal does not saturate the camera sensor.
- In the 'results' tab, the result is displayed in the upper part. Save a tab-separated *.txt file of the result table by clicking on "Save the Result Table".
NOTE: This table consists of the time in seconds and the normalized calcium-dependent fluorescence (corrected by the calcium-independent fluorescence). If previously defined, it also contains the total distance between the TRN and the actuator in µm and the distance between the TRN and the actuator perpendicular to the channel wall in µm. Representative results of the fluorescence intensity increase after stimulation versus the distance of the cell body to the closest actuator are plotted in Figure 4. - If there are multiple neurons in one recording (separated by more than 10 pixels), click on the "Define ROIs" tab again and return to step 4.6.2 with the second neuron.
- Click on the "Help" tab and read about the requirements of the software and how to perform the analysis. To begin, select the "Open video" tab. Specify the video file location for the analyzer: click the "Open a Video" button, navigate to the location of the video, and open it.
SU-8 Lithography and Chip Bonding
The lithography protocol and PDMS molding follow standard procedures. Details can be found elsewhere23,24,25,26. The PDMS should peel off the wafer without problems after curing. If the SU-8 features rip off during PDMS peeling, either the SU-8 adhesion layer or the silanization was insufficient. If plasma-activation of the glass coverslips and the PDMS chips is not sufficient, weak bonding can result in leakage out of the trapping channel or pneumatic actuators resulting in performance degradation and absence of stimulation. If fluid leakage is evident, the chip is unusable.
Actuator Performance
All of the PDMS actuators and the tubing connecting the chip to the air reservoir must be properly sealed to ensure proper pneumatic actuation of the diaphragms. Repeated insertion and removal of the PE tubing (plumbing) will damage PDMS holes and lead to a decrease in pressure. Such a loss of pressure will limit actuator deflection and the force applied to the skin of the immobilized animal. Deflection of the diaphragm can be measured using an empty chip (no animal present) by performing several actuation cycles with the desired pressure and comparing the quantitative values with theoretical predictions. The deflection of the diaphragm was not measured in presence of an animal in the trapping channel, because the membrane and worm boundary are difficult to visualize. This makes precision calibration with a trapped animal not possible. Table 1 lists the expected deflection as a function of pressure. These values were calculated using elastic plate theory and details can be found in Nekimken et al.23 This relationship is expected to vary with differences in diaphragm thickness generated during chip casting and other factors such as the integrity of the bonding between the chip and the glass and seals connecting the chip to the pressure sources. Here, values were reproducible with slight variation not exceeding 1 µm at 450 kPa of pressure. We have characterized the performance of the diaphragms for all three different stimulus profiles and refer the interested reader to Nekimken et al.23 In summary, there are several factors to consider if the values deviate from Table 1: 1) a leak in the tubing or tubing connectors, 2) the PDMS has a different elasticity, which can result from an alternative ratio of base polymer and curing agent, or 3) the PDMS has aged and continued to crosslink over time. Therefore, use of chips which have been prepared within a month of their application is recommended.
Trapping Performance
After inserting the worms into the inlet of the chip, a gentle pressure with the syringe should place one single animal inside the trapping channel and present its skin alongside the six actuators (Figure 1). Importantly, the nose of the animal does not necessarily need to protrude into the buffer reservoir. Instead, it is more important to position the animal such that the neurite is adjacent to the nearest actuator and its cell body is within the field of view of the microscope. To achieve optimal neuronal activation, the distance between the actuator and cell body should be adjusted such that the actuator lies anterior to the cell body of interest. In our experience, for neurons stimulated posterior to the cell body, no activation will take place and calcium transients will be absent. Proper immobilization can be achieved with young adult or one-day old hermaphrodites (devices optimized for these two ages are available on the same mask). Smaller animals will slip into the collection reservoir or move laterally inside the trapping channel. Animals that are too large will be squeezed into the channel, which might result in premature activation of its TRNs and subsequent failure to detect mechanoreceptor transients. Also, removal of large worms is challenging, leading to death of the animal and/or clogging of the device. With this design, the PLM neuron usually cannot be immobilized completely, so it is free to move laterally and vertically. Although we have successfully activated PLM neurons, recording calcium transients in these neurons is difficult due to vertical movement of the tail. Another trapping design has been used to record from PLM33.
Imaging of Calcium Transients and Analysis
Once in place, the animals can be stimulated using one of the six actuators. The six actuators are positioned to deliver mechanical stimuli to each of the six TRNs. A microfluidic flow controller connected to a 450-kPa in-house pressure source was used to deliver to the animal a stimulus protocol consisting of a 2-s 275 kPa step, a 2-s 0–275 kPa ramp, and a 2-s buzz (a 75 kPa, 10 Hz sine superimposed with a 275 kPa step), each separated by a 10-s waiting period. The simultaneously recorded image sequences of the calcium transients were analyzed in Fiji, using a custom-written software available on our GitHub account (see above and https://github.com/HFehlauer/Poking-Analyzer). We found that pressure ramps and pressure steps activated TRNs only if the stimuli achieved pressures above 400 kPa. By contrast, a robust activation of all TRNs stimulated using a sinusoidal, 10-Hz buzz at pressures < 275 kPa was observed (Figure 4). In general, TRNs responded better to a brief sinusoidal stimulus (called 'buzz'), in agreement with previous reports using different methodologies1,9. The stimulation with a buzz is highly efficient, leading to ~ 90% of activation in all neurons and trials tested (Figure 4). Surprisingly, there was no strong correlation observed on whether the stimulus was applied to the dorsal or the ventral side, and was independent of the distance between the cell body and the stimulus position on the neurite. Future studies will have to investigate whether the delay of the maximum response correlates with the stimulus distance.
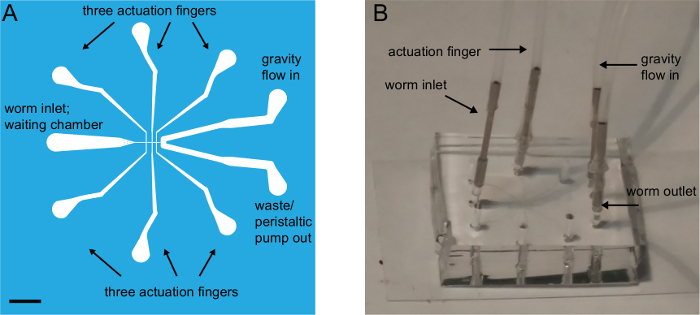
Figure 1: Overview of the chip design. (A) Schematic layout of the photomask and the resulting chip with all connectors indicated. There are six actuation fingers, three on each side of the central trapping channel. Scale bar = 3 mm. (B) Photograph of the microfluidic device with PDMS bonded to the coverslip and connected to tubing in the worm inlet and the worm outlets. Only one of the actuation fingers is connected to the pressure source, while the other five are not occupied. Parts of the figure have been reproduced from reference23 under creative common licenses. Please click here to view a larger version of this figure.
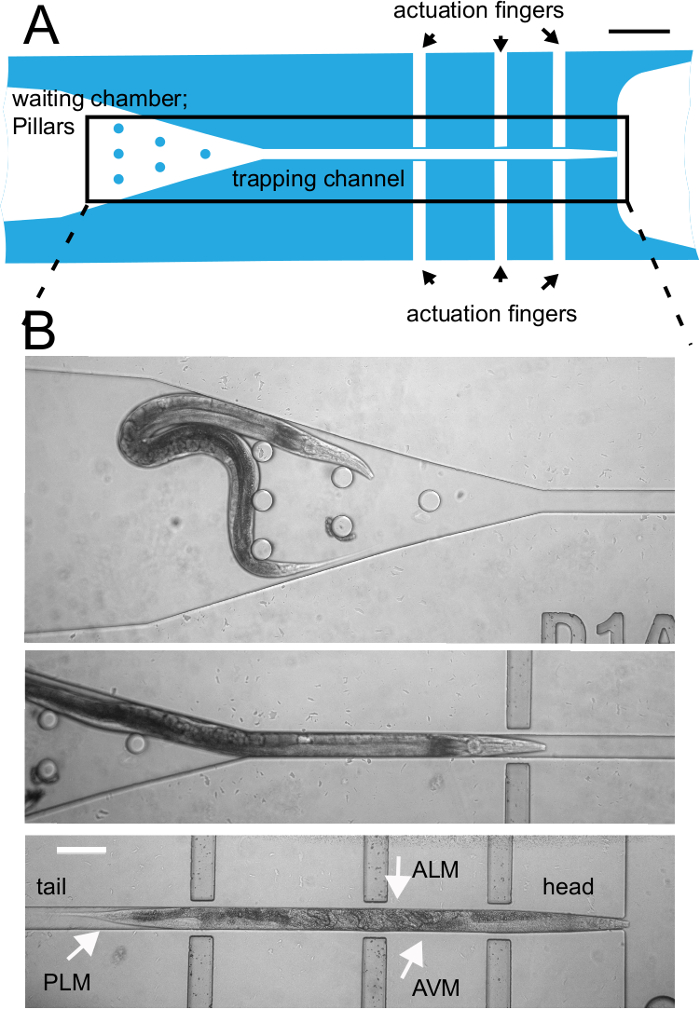
Figure 2: Close-up of the central trapping channel and trapping procedure. (A) Schematic drawing of the trapping channel. The pillars at the worm inlet side help to orient the worm and facilitate loading of the animals. The actuation fingers can be pressurized and indent a thin diaphragm into the trapped specimen. Scale bar = 200 µm. (B) Representative video frame of a worm being trapped in the horizontal channel. The channel is designed such that the worm comes to lay across the six pneumatic actuators. Tail is to the left, head is to the right. Location of individual touch receptor neurons (TRNs) are indicated by arrows and labeled in the figure. Scale bar = 100 µm (in all panels). Parts of the figure have been reproduced from reference23 under creative common licenses. Please click here to view a larger version of this figure.
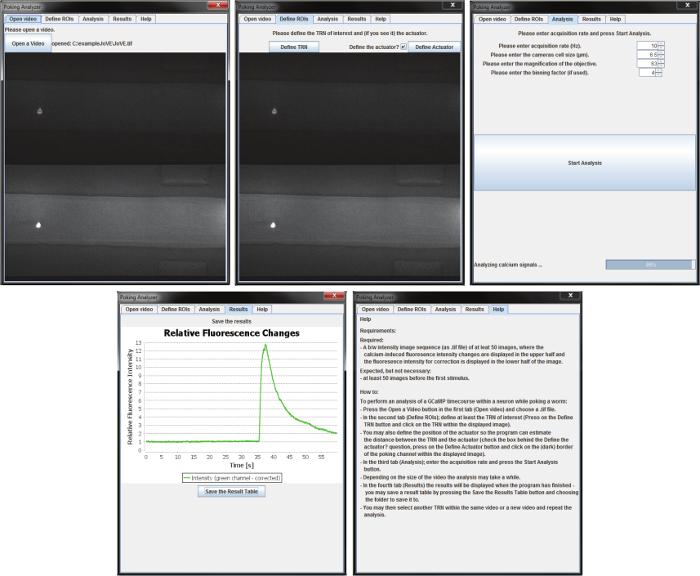
Figure 3: Overview of the provided Fiji plugin to analyze calcium traces acquired with this protocol. The software starts a graphical user interface. The first tab ("Open video") of the user interface shows the button to open a video, it displays the path and the first frame of the opened video. The second tab ("Define ROIs") includes the button to define a touch receptor neuron, a checkbox for activating the button "Define Actuator" and the button "Define Actuator". In the lower part, the first frame of the video is displayed. In the upper part of the third tab ("Analysis"), adjustable parameters for the image analysis are displayed. The middle part shows the "Start Analysis" button. In the lower part, a progress bar shows the progress of the current analysis. In the fourth tab, "Results", the result is displayed as a graph of the relative fluorescence intensity over time. In the lower part, there is a button to save the result table. In the fifth tab, "Help", a help text is displayed showing the requirements for the analysis software and instruction on how to use it. Figure adapted from reference23 under creative common licenses. Please click here to view a larger version of this figure.
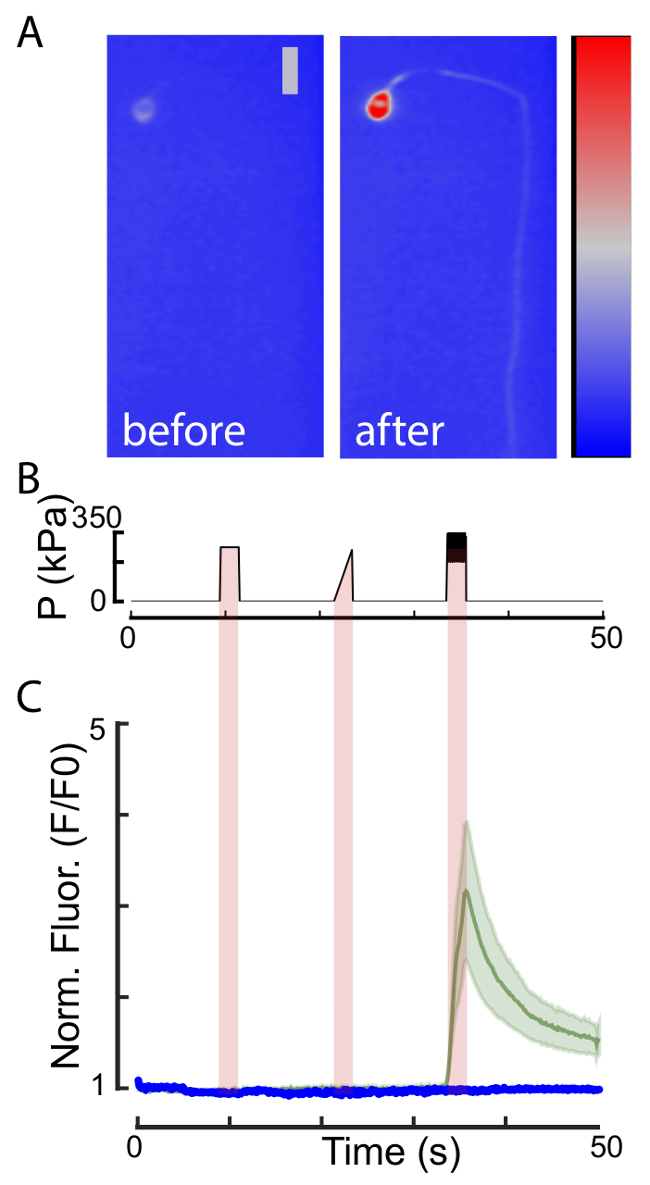
Figure 4: Individual neurons respond to mechanical stimuli. (A) Representative fluorescence micrograph of GCamP6s labeled AVM touch receptor neuron before and after mechanical stimulation with a buzz. Scale bar = 10 µm. Color scale = 1,500-3,500 gray values. (B) Stimulus protocol including 2 s diaphragm excitation representing a 275 kPa step, a 275 kPa ramp, and a sine (75 kPa; 10 Hz) superimposed with a 275 kPa step (buzz). (C) Normalized GCamP6s intensity trace (mean ± SEM as shaded area) recorded from AVM stimulated with the profile shown in B of wild type animals (green, n = 14) and animals mutant in the mechanoreceptor channel subunit mec-4 (blue, n = 10), that is known to facilitate responses to mechanical stress. This indicates that the transients are induced by the applied mechanical stimuli. Parts of the figure have been reproduced from reference23 under creative common licenses. Please click here to view a larger version of this figure.
| Pressure (kPa) | Mean Deflection (µm) | SD | Expected Deflection (µm) |
| 0 | -0.01 | 0.01 | 0 |
| 50 | 0.77 | 0.26 | 0.607143 |
| 100 | 1.57 | 0.58 | 1.21429 |
| 150 | 2.32 | 0.65 | 1.82143 |
| 200 | 3.13 | 0.74 | 2.42857 |
| 250 | 3.86 | 0.79 | 3.03571 |
| 300 | 4.61 | 0.79 | 3.64286 |
| 350 | 5.3 | 0.82 | 4.25 |
| 400 | 5.92 | 0.82 | 4.85714 |
| 450 | 6.43 | 0.75 | 5.46429 |
Table 1: Expected experimental deflection values for applied pressures ranging from 0–450 kPa and their theoretical predictions. Data derived for a step stimulus. Mean ± SD. These data were acquired with an empty trapping channel.
