Using the method outlined here, the goal of this experiment is to study the functional effects of introducing indel mutations to the endogenous Calr locus on hematopoietic cell transformation. The CRISPR/Cas9 system is used as a tool to create endogenous Calr mutations in Ba/F3 cells. Two sgRNAs were chosen to target exon 9 of Calr (Figure 1), in the region where insertions and/or deletion (indel) mutations typically occur in CALR-mutant MPN patients17,18. The first sgRNA (m1) was chosen based on its high cleavage efficiency and favorable off-target scores (Table 1). The second sgRNA (m2) was chosen primarily for its location within exon 9 and for lack of additional sgRNAs in the region to edit with high cleavage efficiency and favorable off-target scores (Table 1). Two distinct sgRNAs (m1 or m2) were used in separate infections to ensure that the observed effects were due to on-target gene editing. Off-target effects are unlikely to be shared by multiple independent sgRNAs. The non-targeting control (scramble) was also used as a negative control. Recruitment of the Cas9 endonuclease to Calr exon 9 is predicted to create DSBs at this locus. The DSBs would then be repaired by NHEJ, which can generate indels of variable sizes (Figure 1).
To develop the in vitro CRISPR/Cas9 system in Ba/F3 cells, cells stably expressing the Cas9 protein were made by lentiviral-mediated transduction as per the protocol described above. Stable Cas9 expression results in robust Cas9 activity. Cas9 activity in Ba/F3-Cas9 cells was measured by pXPR-011, a reporter construct that contains both GFP and a guide targeting GFP15. Cells containing active Cas9 will result in a reduction of GFP15. GFP was measured in the cells using flow cytometry. Ba/F3-Cas9 cells displayed a reduction of approximately 76% in GFP, corresponding to robust Cas9 activity (Figure 2).
Type 1 cytokine receptors, such as the thrombopoietin receptor (MPL), the erythropoietin receptor (EPOR) and the granulocyte colony-stimulating factor receptor (G-CSFR) were each individually, stably expressed in Ba/F3-Cas9 cells to determine their cooperativity with mutant calreticulin in inducing transformation of Ba/F3 cells. Transduction of the sgRNA constructs (m1, m2 or a scramble (non-targeting guide)) was then carried out in each of the Ba/F3 cell lines stably expressing Cas9 and the receptor of interest. Cells were then selected for 7 days with puromycin to allow for sufficient time for CRISPR/Cas9 gene editing. A positive selection pressure was then applied by starving the cells of cytokine (mIL-3). The goal of this starvation pressure is to identify if indels in Calr exon 9, similar to those observed in MPN patients, are selected for, resulting in cytokine-independent growth and transformation of Ba/F3 cells. The growth curve was carried out for a total of 8 days and the cells were counted every 2 days to measure their transformation (Figure 3)19. Cell pellets for genomic DNA extraction were collected before the start of the growth curve and at the end of the growth curve to check for on-target editing and to monitor the indels that expanded post cytokine starvation (Figure 3)19.
Cytokine independent growth was observed in Calr-targeted Ba/F3-MPL-Cas9 cells (Figure 4B), but not Calr-targeted parental Ba/F3-Cas9 cells (Figure 4A), or Ba/F3-Cas9 cells ectopically expressing EPOR (Figure 4C) or G-CSFR (Figure 4D)19. To confirm that mIL-3 independent growth in Calr-targeted Ba/F3-MPL-Cas9 cells was a result of on-target gene editing, cells were harvested 8 days post mIL-3 withdrawal and genomic DNA was extracted19. A 422 bp region spanning the target site was amplified using the primers listed in Table 3. Sub-cloning of PCR amplicons was performed and 30 individual clones were sent for Sanger sequencing. For m1 or m2 Calr-targeted Ba/F3-MPL-Cas9 cells, all sub-clones contained indels of varying sizes at the intended cut site (Figure 5)19. 11 out of 13 m1 sub-clones were found to contain indels that led to +1 bp frameshift mutations (Figure 5), similar to those found in patients. For m2 Calr-targeted cells, 10 out of 17 sub-clones were found to contain indels that led to +1 bp frameshifts (Figure 5)19. These data confirm that CRISPR/Cas9- mediated introduction of +1bp frameshift mutations to the endogenous Calr exon 9 locus is sufficient to confer oncogenic activity to Calr19. The sequencing data in Figure 5 also suggests that the introduction of heterozygous +1bp frameshift mutations to the endogenous Calr locus is sufficient to transform Ba/F3-MPL cells, consistent with the observation that CALR mutations are typically heterozygous in MPN patients17,18. Since NHEJ repair results in high heterogeneity between the indels, single cell sorting to isolate a clone of interest could be performed. This would allow the researcher to study the effects of specific mutations created by CRISPR/Cas9.
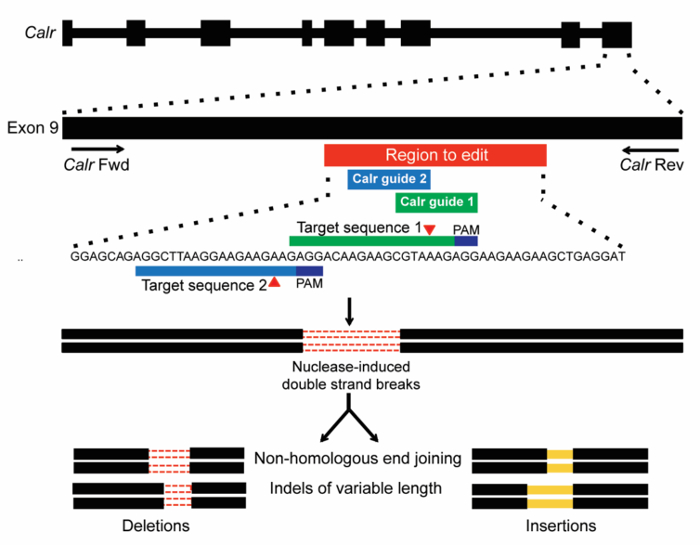
Figure 1: Schema of CRISPR/Cas9 gene-editing targeting of exon 9 of Calr. Two sgRNAs were designed to target a site within the mouse Calr exon 9 region corresponding to that targeted by the +1bp frameshift mutations in human MPN (red bar). Target sequences with PAMs (dark blue) are shown, and expected sites of cleavage by Cas9 are indicated by red triangles. DSBs generated by CRISPR/Cas9 are then repaired by NHEJ, which is expected to generate indels of variable length19. Please click here to view a larger version of this figure.
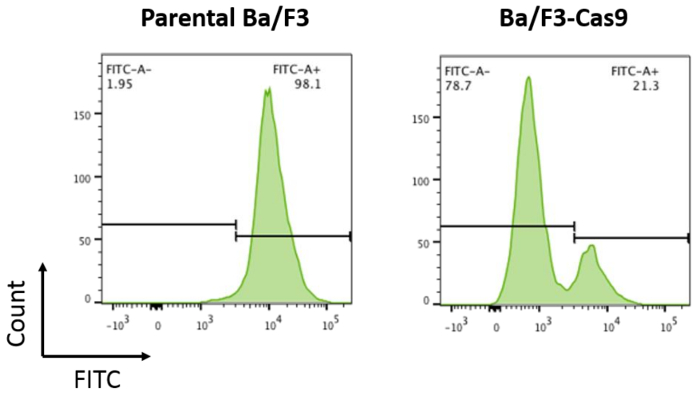
Figure 2: Cas9 activity assay. Parental Ba/F3 cells and Ba/F3 cells overexpressing Cas9 were transduced with pXPR-011 to measure Cas9 activity. A reduction in GFP correlates with increased Cas9 activity. Parental Ba/F3 cells are ~100% FITC+ compared to Ba/F3-Cas9 cells where the FITC is reduced by ~76%. Please click here to view a larger version of this figure.
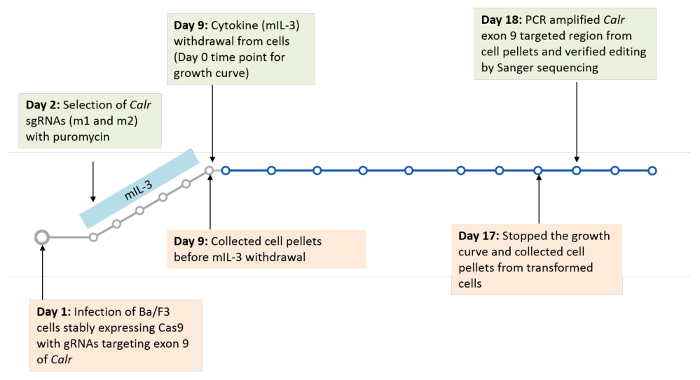
Figure 3: Timeline for infection and selection of sgRNAs in Ba/F3 cells. Please click here to view a larger version of this figure.
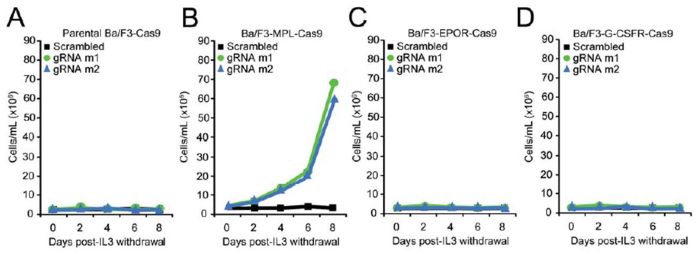
Figure 4: Introduction of +1 bp frameshift mutations into the endogenous Calr locus is sufficient to confer oncogenic activity to Calr. (A-D) Growth curves in parental Ba/F3-Cas9 cells (A) Ba/F3-MPL-Cas9 cells (B), Ba/F3-EPOR-Cas9 cells (C), and Ba/F3-G-CSFR-Cas9 cells (D) demonstrates IL-3 independent growth in Calr-targeted Ba/F3-MPL-Cas9 cells only19. Please click here to view a larger version of this figure.

Figure 5: Sanger sequencing verification. Confirmation of on-target editing of endogenous Calr (exon 9) in Ba/F3-MPL cells. +1 bp frameshift mutations are indicated in black19. Please click here to view a larger version of this figure.
| Target gene | sgRNA ID | Target sequence (5' to 3') | Strand | Cleavage Efficiency | Off-target score | ||
| Calr | m1 | AGAGGACAAGAAGCGTAAAGAGG | + | 0.7 | 70 | ||
| Calr | m2 | GAGGCTTAAGGAAGAAGAAGAGG | + | 0.3 | 28 | ||
Table 1: 20-mer protospacer sequences including the PAM site (in bold) for two sgRNAs targeting exon 9 of Calr19.
| Primer ID | Sequence (5' to 3') |
| m1-F | CACCGAGAGGACAAGAAGCGTAAAG |
| m1-R | AAACCTTTACGCTTCTTGTCCTCTC |
| m2-F | CACCGAGGCTTAAGGAAGAAGAAG |
| m2-R | AAACCTTCTTCTTCCTTAACCTC |
Table 2: Protospacer sequences and their reverse complements with "CACC" and "AAAC" added for cloning into pLenti-Guide vector using BsmBI restriction enzyme19.
| Primer ID | Sequence (5' to 3') |
| Calr_Fwd | ACCACCTGTCTTTCCGTTCT |
| Calr_Rev | GGCCTCTACAGCTCATCCTT |
Table 3: CRISPR screening PCR primers upstream and downstream of the sgRNA cleavage site.
