The SiC-seq experimental workflow contains 3 PDMS microfluidic devices fabricated using a soft lithography procedure (Figure 1). A co-flow dropmaker (Figure 3A) generates 25 µm of digital barcode droplets for labeling genomic DNA with a unique single-cell identifier. The barcode oligonucleotides consist of a 15 bp degenerate sequence flanked by PCR handles for amplification (Table 2, BAR primer). The barcodes are diluted to a femtomolar concentration to achieve the single-molecule encapsulation, and all droplets receive either 0 or 1 barcode fragment(s). The droplets containing a barcode are amplified, yielding many copies of double-stranded barcode amplicons. A nucleic acid stain is used to verify the successful amplification and quantify the encapsulation rate of the barcode fragments (Figure 4B). The microgels are generated by co-flowing a bacterial cell suspension and a molten agarose gel at equal flow rates (Figure 2A). The agarose is prepared at twice the desired final concentration, as the co-flow dropmaking process effectively dilutes the aqueous solutions by a factor of 2. As the agarose cools, it solidifies into a 25 µm diameter microgel occupying the spherical volume of the droplet.
A series of wash and lysis steps purifies the high-molecular-weight genomic DNA in the microgels (Figure 2B). After breaking the emulsions, aqueous washes are carried out in large volumes to dilute out trace organic solvents which can inhibit the downstream enzymatic treatments. The washed microgels are observed under a microscope to verify the cell encapsulation rate (Figure 4A). A cocktail of enzymes with broad lytic activity is added to the microgel suspension to digest the cell walls of the bacteria and eukaryotic microbes19. A second treatment with Proteinase K and detergent degrades the proteins and solubilizes cell debris.
Tagmentation of the purified DNA is carried out in droplets to avoid potential cross-contamination resulting from the diffusion of small tagmented DNA fragments between the microgels18. A droplet encapsulation device (Figure 3B) compartmentalizes each microgel with a buffer and tagmentation enzyme, which simultaneously fragments double-stranded DNA while also "tagging" it with a preloaded oligonucleotide20. The microgels are loaded into the droplets as close-packed particles, achieving encapsulation rates approaching 1 microgel for every drop with few doublets21 (Figure 4C).
In the final step of the microfluidic workflow (Figure 2C), a device performs a double merger operation combining 1 barcode drop, 1 microgel-containing drop, and the amplification mix in a controlled two-step process. First, a droplet containing PCR reagent is paired and merged with a barcode drop in the region shown in yellow (Figure 5). Saltwater electrodes in the microfluidic channel produce a high electric field gradient which triggers the droplet merger. In a similar manner, the first merged droplet is paired with a microgel droplet and merged a second time in the region shown in red. The droplets are collected and thermal cycled off-chip in a single-overlap extension (SOE) PCR. The overlapping complementary ends of the barcode and the tagmented genomic DNA allow fusion and exponential amplification of only properly barcoded constructs.
The sequencing data is first filtered by a read quality and then parsed by grouping the reads according to their 15-bp single-cell barcode sequence. For a barcode group to be considered valid, it should contain a minimum number of reads; this thresholding limits the analysis to cells with a useful amount of sequencing data and removes the PCR-mutated barcode "orphans" from the dataset. In this sample run, the minimum is set to 7.5 kbps per group (50 reads of 150 bp each). A histogram of the barcode counts versus the group size shows that a significant portion of the valid barcode groups is just above the threshold size (Figure 6A).
In a control experiment where the microbial community composition is known, the purity and relative abundance metrics are used to evaluate the quality of a SiC-seq run. Here, a synthetic 10-cell community consisting of 3 gram-negative bacteria, 5 gram-positive bacteria, and 2 yeasts is analyzed. The purity of a given barcode group is defined as the number of reads mapping to the most common genome in the group divided by the total number of reads in the group. The vast majority of the barcode groups have purities greater than 0.95 (Figure 6B). Relative abundance of the cell types is calculated by counting the raw reads and by counting the barcode groups, where the groups are assigned a cell type corresponding to the consensus of its member reads (Figure 6C). The abundance of reads and barcode groups track in roughly equal proportions, indicating that the cell populations are being sampled such that certain species are not contained in disproportionately small or large barcode groups. Plotting the aggregate coverage of all barcode groups from a single species indicates a high coverage across the entire genome, with few or no dropout regions (Figure 7). The uniformity of coverage can be verified with a frequency distribution of normalized coverage values, with most values centered around the average (Figure 7, inset).
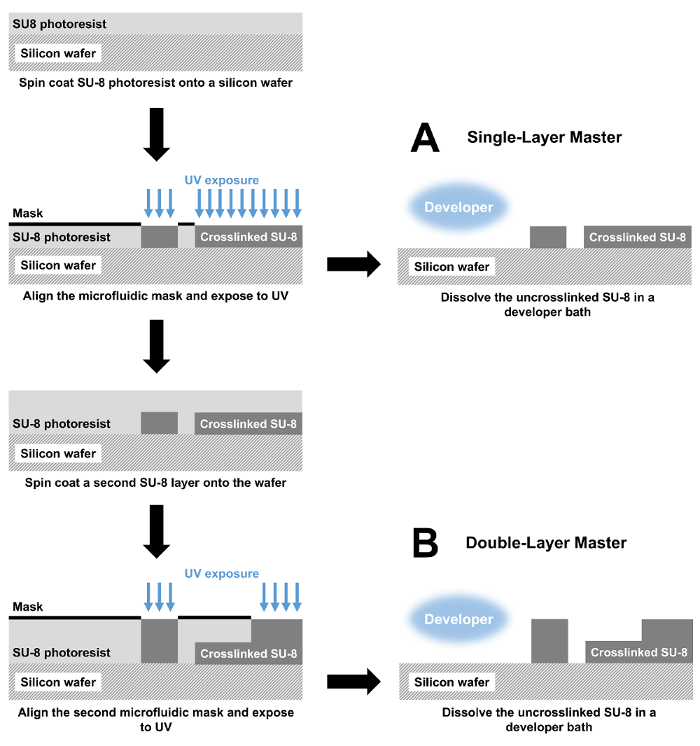
Figure 1: Fabrication of microfluidic devices by photolithography. (A) Master molds with a single feature height are fabricated by spin coating a layer of SU-8 photoresist onto a silicon wafer. The photoresist is then patterned with a photolithographic mask and UV light, crosslinking the exposed SU-8. Finally, uncrosslinked SU-8 is dissolved in a developer bath. The resulting mold is used to cast PDMS which is bonded to a glass slide to produce the complete microfluidic device. (B) For a double-layer device, the fabrication similarly begins with spin coating and exposure steps. These steps are then repeated to create a two-layer device. Please click here to view a larger version of this figure.
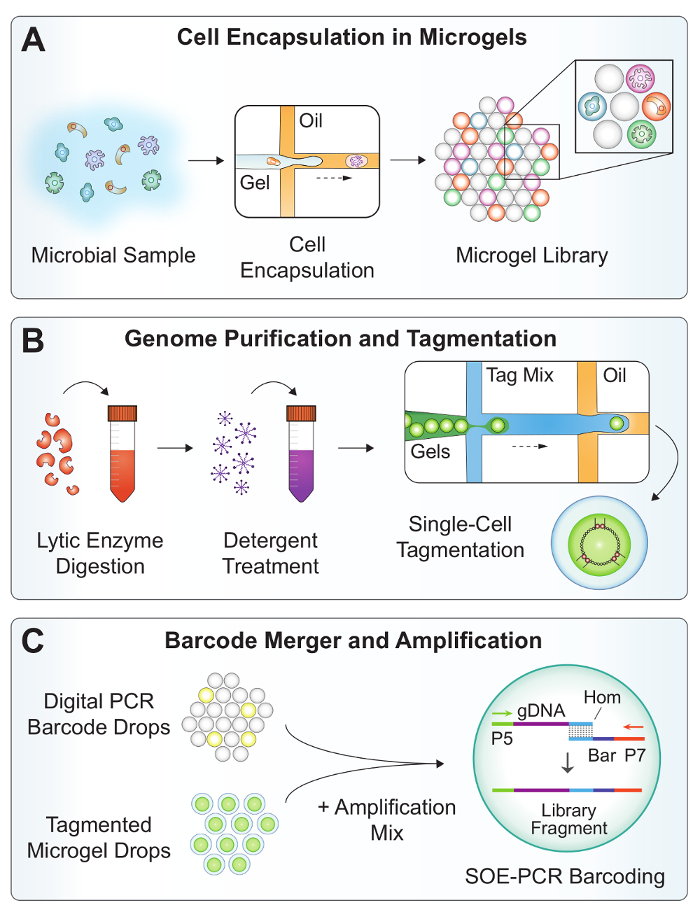
Figure 2: Overview of the SiC-seq workflow. (A) A microbial suspension is co-flowed with molten agarose in a dropmaker device to encapsulate single cells in microgels. (B) The microgels are subjected to a series of washes to purify the bacterial genomic DNA. Lytic enzymes digest the cell walls of gram-positive bacteria and yeasts, and detergent solubilizes the cellular debris. The microgels are re-encapsulated into droplets for the tagmentation to reduce cross-contamination. (C) The microfluidic merger combines a digital PCR barcode, a tagmented microgel genome, and an amplification mix at a rate >1 kHz. Off-chip SOE-PCR splices a unique single-cell barcode onto the tagmented genome and selectively amplifies fully barcoded constructs. Please click here to view a larger version of this figure.
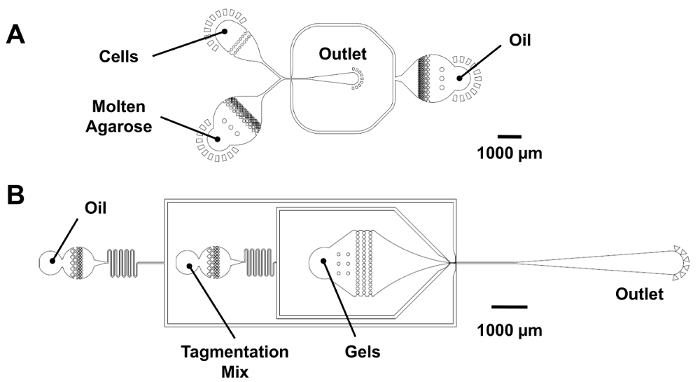
Figure 3: Microfluidic devices for dropmaking and microgel re-encapsulation. (A) This panel shows a co-flow dropmaker (25 µm of feature height). Cells and molten agarose are introduced into the device at equal flow rates to produce 25 µm droplets at a 25 µm x 25 µm junction. For the digital barcode dropmaking, the cell inlet is plugged, and a PCR mix is introduced into the agarose inlet. (B) This panel shows a microgel re-encapsulation device (25 µm of feature height). The microgels flow into a funnel-shaped inlet to maintain their close-packed ordering and receive a volume of tagmentation mix prior to the re-encapsulation at a 25 µm x 30 µm junction. Please click here to view a larger version of this figure.

Figure 4: Micrographs of droplets and microgels. (A) This panel shows washed 25 µm microgels prior to enzymatic lysis. The bacteria are fluorescently stained for the quantification of the encapsulation rate. Poisson loading statistics dictate that the cells should be encapsulated at a rate of 1 in 10 drops or less to minimize the frequency of multiple-encapsulation events. (B) This panel shows a fluorescence microscopy image of 25 µm digital barcode droplets treated with a nucleic acid stain. The droplets containing amplified barcode fragments produce a strong fluorescence signal. (C) This panel shows microgels re-encapsulated in 50 µm drops. The close-packing of the microgels allows for encapsulation rates approaching 1 gel per drop with few doublets. Please click here to view a larger version of this figure.
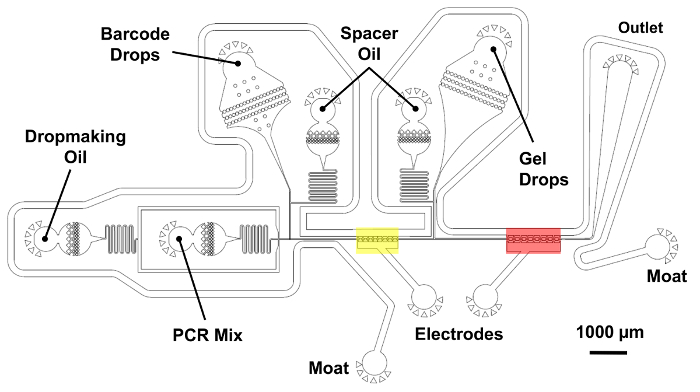
Figure 5: Microfluidic double merger device for single-cell genome barcoding. A two-step merger operation pairs barcode droplets with tagmented genomes at a high-throughput. A droplet of PCR mix is first generated and merged with a barcode droplet in the region shown in yellow using saltwater electrodes. Next, a droplet containing a microgel is introduced and merged a second time in the region shown in red. Oil inlets allow for precise control of the spacing between the reinjected droplets. The barcode reinjection chamber and its spacer oil are placed on the shorter 25 µm layer, shaded in blue. All other device features belong to the thicker layer with 45 µm of total height. Please click here to view a larger version of this figure.
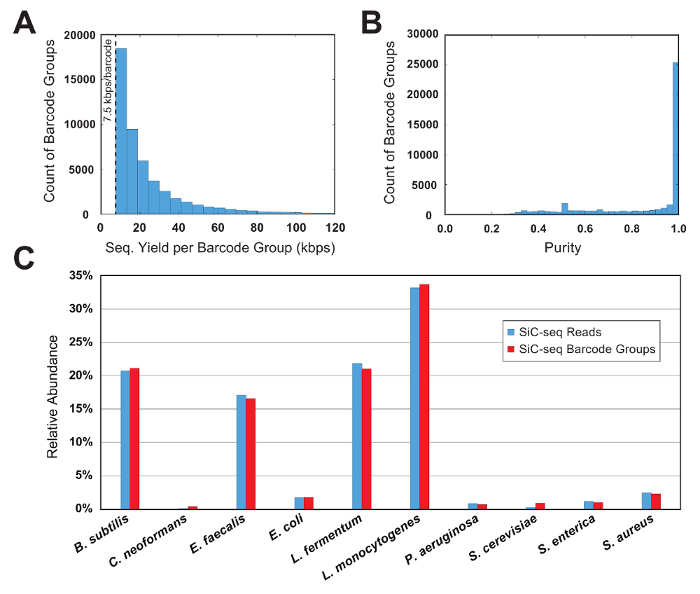
Figure 6: Barcode group metrics for a 10-cell synthetic microbial community. (A) This panel shows the distribution of the barcode group sizes. The number of groups of a given size decreases exponentially as the group size increases. A minimum threshold of 7.5 kbps per group limits the analysis to groups with a sufficient amount of information and removes the PCR-mutated sequence "orphans." (B) This panel shows the distribution of barcode group purities. The vast majority (>90%) of groups are of very high purity (>95%). (C) This panel shows the relative abundance of 10 species calculated at the read and barcode group level. The 2 counting methods produce similar results, indicating that the barcode group sizes are consistent across species. Please click here to view a larger version of this figure.
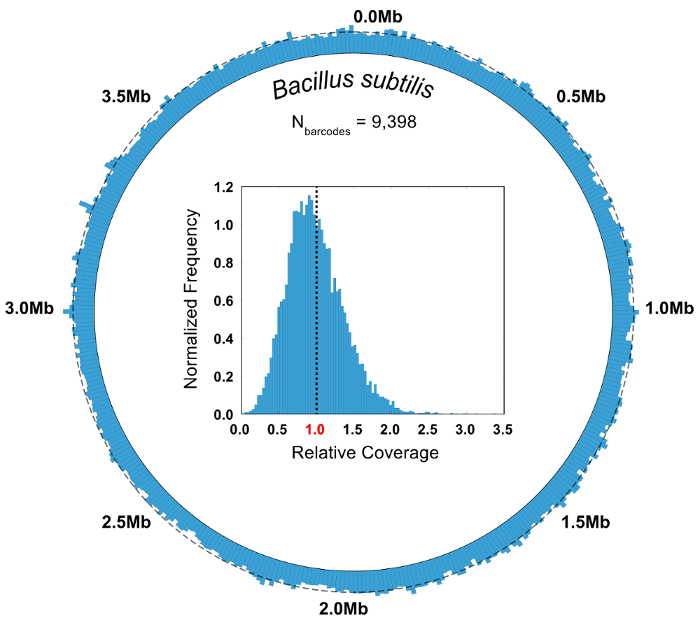
Figure 7: Aggregate genomic coverage of Bacillus subtilis barcode groups. The reads from all the barcode groups mapping to the bacterium B. subtilis (N = 9,398) are pooled and analyzed in aggregate. A circular coverage map illustrates the coverage uniformity of the SiC-seq reads, with no observable dropout regions. A dashed line around the circumference indicates the average coverage (5.55x). The inset histogram of the relative coverage frequencies shows that a bulk of the bases are covered at a depth near the genome-wide average, represented by the dashed line. Please click here to view a larger version of this figure.
| Device | 1st Layer Height (µm) | 1st Layer Spin Speed (rpm) | 2nd Layer Height (µm) | 2nd Layer Spin Speed (rpm) |
| Co-flow dropmaker | 25 | 4000 | N/A | N/A |
| Gel re-encapsulator | 25 | 4000 | N/A | N/A |
| Double merger | 25 | 4000 | 20 | 5000 |
Table 1: Microfluidic device fabrication parameters. This table shows a listing of the microfluidic devices used in the SiC-seq workflow with their required speeds for photoresist spin coating (based on the manufacturer's specifications for SU-8 3025).
| Label | Sequence (5' > 3') | ||||
| BAR | GCAGCTGGCGTAATAGCGAGTACAATCTGCTCTGATGCCGCATAGNNNNNNNNNNNNNNNTAAGCCAGCCCCGACACT | ||||
| DNA_BAR | CTGTCTCTTATACACATCTCCGAGCCCACGAGACGTGTCGGGGCTGGCTTA | ||||
| P7_BAR | CAAGCAGAAGACGGCATACGAGATCAGCTGGCGTAATAGCG | ||||
| P5_DNA | AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTC | ||||
| I7_READ | GCCCACGAGACGTGTCGGGGCTGGCTTA | ||||
Table 2: Primer sequences.
Supplemental File 1: Please click here to download this file.
Supplemental File 2: Please click here to download this file.
