Figure 2 shows the visualized superficial tangential migration in a flat-mount culture at an elapsed time (0, 9, 18, 27 h) after onset of recording. Movie 1 is a time-lapse movie of 10 min-intervals over a period of 28 h and 50 min. The frame is selected for focusing on the migrating cells from the labeled lower-left corner of the frame to the unlabeled space (Figure 3A). The mass movement of the migrating cells (GFP; upper left panel, Movie 1) and their nuclei (mCherry-Nuc; upper right panel) can be observed with the merged movie (lower panel, Movie 1). Directionality of the cell migration can be examined by focusing on the dispersing cells from the labeled center to all directions (Movie 2, Figure 3B).
The clear images of the nuclear movement (Movie 2; mCherry-Nuc, right panel) allows us to trace the cell nuclear migration by automatic tracking using a Particle Tracker plugin10 of a Fiji image processing application of ImageJ11 (Movie 3, Figure 3B). Temporal changes of the trajectories of the tangential migration can be visualized to prove the dispersing migration in omni-directions.
Individual cell behavior with the sequential morphological change of the leading process, trailing process and nuclei can be manifested with higher magnification images of 5 min-intervals (Movie 4, Figure 3C).
Another type of tangential migration in the middle layers7 can be visualized using a similar protocol (Movie 5, Figure 3D). Bidirectional linear migration along the axon fasciculus running dorsal to ventral (top to down) is evident7.

Figure 1. Protocol flow chart. Step 1: Electroporation in ovo. Step 2: Laying tectal tissue so that the pia side is attached to the insert. Step 3: Inverted fluorescent microscope with laser confocal unit. Please click here to view a larger version of this figure.
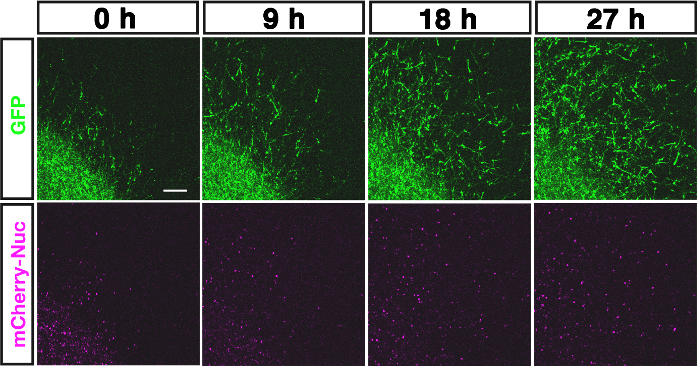
Figure 2. Visualization of superficial tangential migration in flat-mount culture. The tangentially migrating cells (GFP; upper panel) and the nucleus (mCherry-Nuc; lower panel) in the superficial layers of the optic tectum are shown at 0, 9, 18, 27 h after onset of the culture from E7.0. The cells at the lower-left corner of the frame are labeled at 0 h (see Figure 3A). Scale bar: 100 µm. Please click here to view a larger version of this figure.
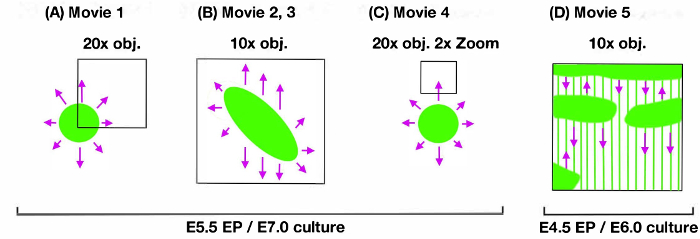
Figure 3. Schematic figure illustrating the mount conditions for time-lapse imaging. The shape of the fluorescent labeling (green), initial direction of cell migration (magenta), and video frame (black square) were illustrated with magnification of the objective lens (obj) and digital zoom (zoom) in the upper field, with day of electroporation (EP) and onset of culture (culture) in the bottom field. (A) Movie 1, (B) Movie 2 and 3, (C) Movie 4, (D) Movie 5. Please click here to view a larger version of this figure.
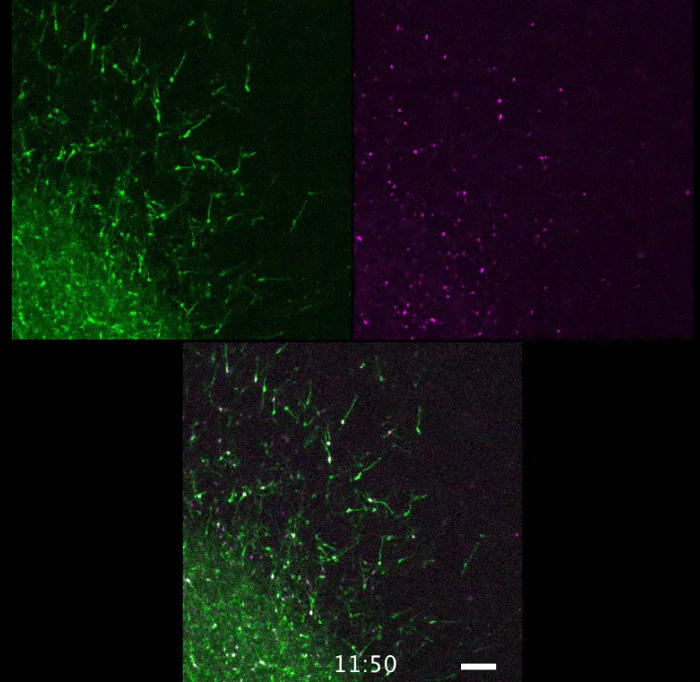
Movie 1. Visualization of superficial tangential migration in a flat-mount culture. Movement of the tangentially migrating cells (GFP; upper left panel) and their nuclei (mCherry-Nuc; upper right panel) in the superficial layers of the optic tectum after onset of the culture from E7.0. The merged image is shown in the lower panel. The cells at the lower-left corner of the frame are labeled at 0 h (see Figure 3A), and the time-lapse images were captured over 28 h and 50 min. Scale bar: 100 µm. Please click here to view this video. Right-click to download.
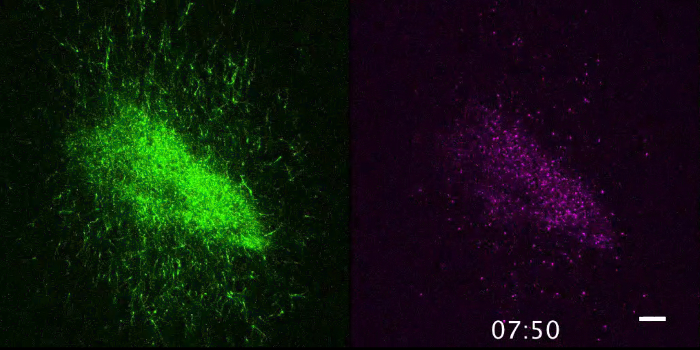
Movie 2. Dispersing movement of the tangentially migrating cells (GFP; left panel) and their nuclei (mCherry-Nuc; right panel) from the center of both panels are shown over 48 h (see Figure 3B). Scale bar: 100 µm. Please click here to view this video. Right-click to download.
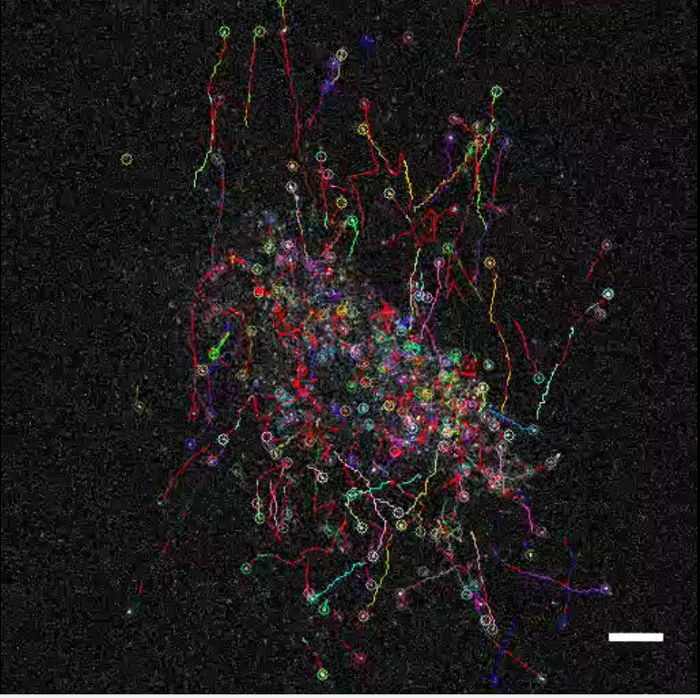
Movie 3. Trajectories of tangential migration. Displacement of the cell nucleus (the right panel of Movie 2) was tracked to visualize the trajectories of the tangential migration. Scale bar; 100 µm. Please click here to view this video. Right-click to download.
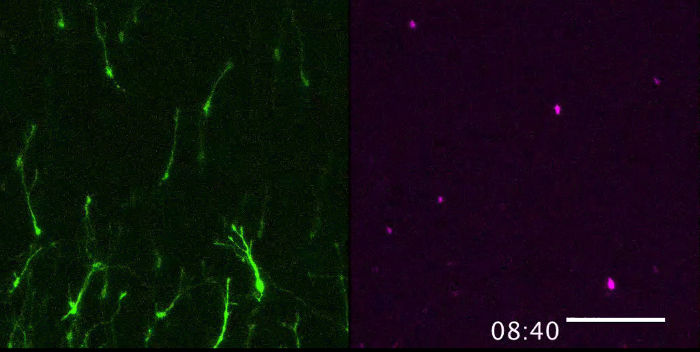
Movie 4. Individual cell behavior in higher magnification. Movement of the individual cells (GFP; left panel) and their nuclei (mCherry-Nuc; right panel) are shown in higher magnification over 24 h (see Figure 3C). Branching process of the leading process can be recognized. Scale bar: 100 µm. Please click here to view this video. Right-click to download.
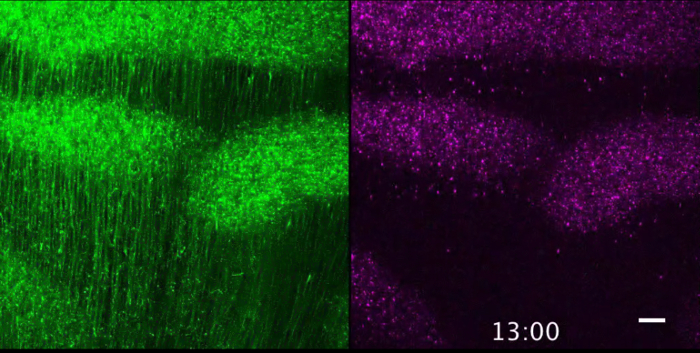
Movie 5. Middle layer migration. Movement of the tangentially migrating cells (GFP; left panel) and their nucleus (mCherry-Nuc; right panel) in the tectal middle layers are shown over 24 h after onset of the culture from E6.0 (see Figure 3D). Linear migration along dorso-ventral axis (top to down) is remarkable. Scale bar: 100 µm. Please click here to view this video. Right-click to download.
