Representative necropsy images of a mouse with a large fluid-filled primary prostate tumor in the anterior prostate region are shown in Figure 2A. In contrast, Figure 2B, shows representative necropsy images of a mouse with a large solid primary prostate tumor for which individual prostate regions are indistinguishable. Fluorescent dissection images show the same solid prostate tumor from Figure 2B expressing GFP, indicating that the tumors cells express Cre (Figure 2C). Tissue that does not express probasin, such as the bladder, express Tomato and thus does not express Cre (Figure 2C). The liver and lungs from the mouse from Figure 2B have metastatic tumors expressing GFP, showing that they originated from the primary prostate tumor, and are surrounded by normal tissue that expresses Tomato (Figure 2C). Finally, the pelvic lymph node from this mouse expresses GFP and not Tomato, indicating that this metastatic tumor has overtaken this organ and no normal tissue remains (Figure 2C).
We show images in Figure 4 of organoids we have generated from a solid prostate tumor. At Day 1, small organoids are forming, as seen in the representative phase contrast images. Fluorescent images on Day 1 show that both Tomato and GFP expressing cells are present in the tumor organoid culture (arrows). However, by Day 7 when prostate tumor organoids have fully formed, these organoids are expressing GFP and not Tomato. These data suggest that these organoids have originated from tumor cells that were expressing Cre and not from normal epithelial cells. These tumor organoids continue to be only GFP-positive as we expand our culture to passage 1 and 2.
In Figure 5, we show images of organoids we have generated from a fluid-filled prostate tumor. On Day 1, small organoids are forming, and fluorescent images show that both Tomato- and GFP-expressing cells are present in the organoid culture — similar to our observation at Day 1 for organoids generated from a solid prostate tumor (Figure 4). However, organoids from a fluid-filled prostate tumor express either GFP or Tomato at Day 7 — indicating that organoids have formed from cells that do not express Cre. This pattern continues at passage 1 and passage 2, where the culture has both Tomato- and GFP-expressing organoids. Further analysis of these organoids is severely limited because the line is a mixture of normal epithelial organoids and tumor organoids. We believe that fluid-filled prostate tumors are suboptimal in generating tumor organoids simply because there is a greater percentage of normal prostate epithelial cells. Since both normal prostate epithelial cells and prostate cancer cells form organoids, the lines generated from fluid-filled prostate tumors are a mixture of normal and cancer organoids. We obtain pure tumor organoid lines from fluid-filled prostate tumors by flow sorting for GFP-positive cells and generating organoids from that population of cells. Solid prostate tumors are primarily comprised of tumor cells, therefore organoids generated from these tumors are a more pure population of cancer organoids without prior sorting for GFP.
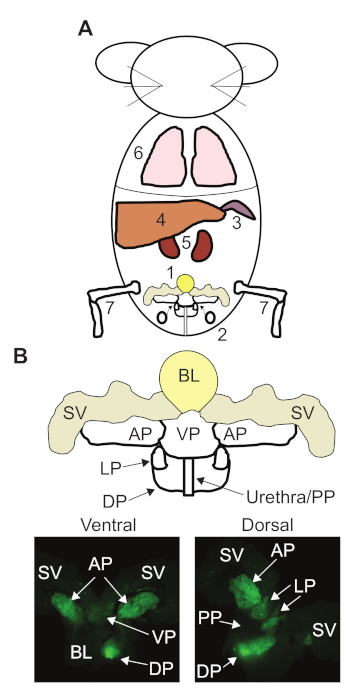
Figure 1: Our recommended dissection order for prostate cancer (PCa) genetically engineered mouse models (GEMMs) and anatomy of the mouse prostate. (A) The order we recommend in our protocol for dissecting the major organs from a PCa GEMM. 1. Urogenital region. 2. Pelvic lymph nodes. 3. Spleen. 4. Liver. 5. Kidneys. 6. Lungs. 7. Tibia and Femur. (B) Map of the mouse urogenital region and prostate anatomy. Fluorescent dissection images of a 12 week old mouse expressing probasin-Cre and the mT/mG Cre reporter transgene. Bladder (BL), seminal vesicles (SV), anterior prostate (AP), ventral prostate (VP), lateral prostate (LP), dorsal prostate (DP), and proximal prostate (PP). Please click here to view a larger version of this figure.
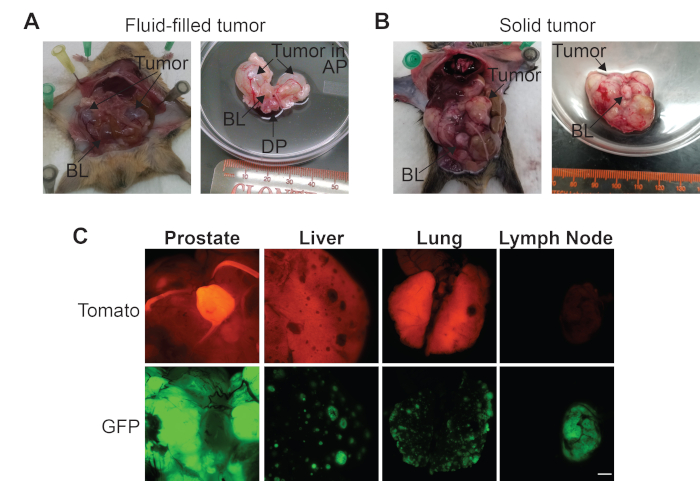
Figure 2: Representative dissection images of prostate cancer (PCa) genetically engineered mouse models (GEMMs). (A) The abdominal cavity prior to removal of the urogenital region and the urogenital region with a fluid-filled prostate tumor. (B) The abdominal cavity prior to removal of the urogenital region and the urogenital region with a solid prostate tumor. (C) Representative Tomato and GFP fluorescent images of a solid prostate tumor, liver, lung, and pelvic lymph node from a PCa GEMM that develops metastatic lesions. Scale bar = 5 mm. Bladder (BL), anterior prostate (AP), and dorsal prostate (DP). Please click here to view a larger version of this figure.
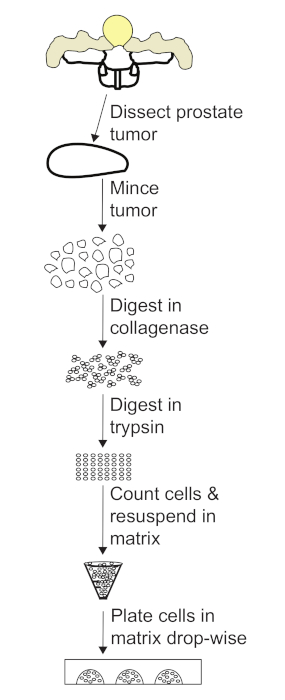
Figure 3: Flow chart of the protocol for generating prostate tumor organoids. After dissecting the prostate tumor, mince the tissue into 1 mm pieces. Digest the tumor pieces in collagenase, collect the cells, and digest in trypsin to obtain a single cell suspension. After counting cells, resuspend in volume of matrix required for a 1.0 x 106 cell/mL cell concentration. Plate domes in dish using a drop-wise method. Please click here to view a larger version of this figure.
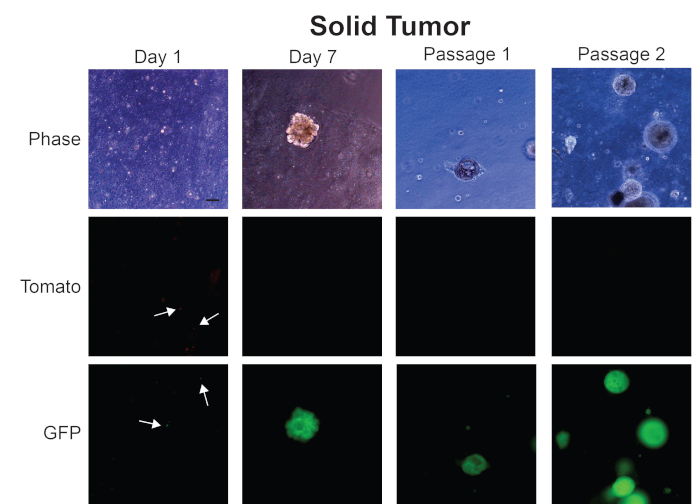
Figure 4: Representative images from generation of mouse prostate tumor organoids from a solid prostate tumor. Representative phase contrast, Tomato, and GFP fluorescent images from Day 1, Day 7, Passage 1, and Passage 2 of organoids generated from a solid mouse prostate tumor. Scale bar = 100 µm. Arrows indicate individual cells in fluorescent images. Please click here to view a larger version of this figure.
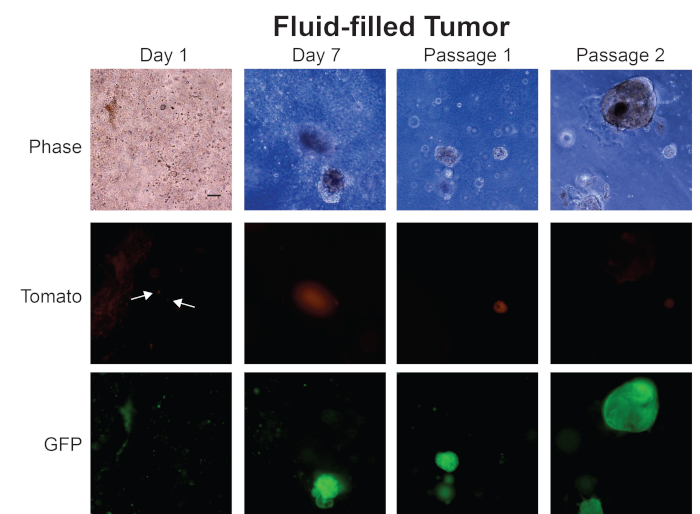
Figure 5: Representative images from generation of mouse prostate tumor organoids from a fluid-filled tumor. Representative phase contrast, Tomato, and GFP fluorescent images from Day 1, Day 7, Passage 1, and Passage 2 of organoids generated from a fluid-filled mouse prostate tumor. Scale bar = 100 µm. Arrows indicate individual cells in fluorescent images. Please click here to view a larger version of this figure.