To enable the conditional expression of Cas9, we developed a dual lentiviral vector construct consisting of a U6-driven promoter to constitutively express sgRNA, and an EF-1α core promoter to drive the expression of the DD-Cas9 fusion protein (Figure 1A)19. As a paradigm to illustrate the robustness and efficiency of the system, we transduced the lung carcinomatous A549 cell line with the lentiviral construct. The levels of Cas9 in the presence or absence of the ligand Shield-1 were measured by reverse transcription-polymerase chain reaction (RT-PCR) and Western blot analysis using an anti-Flag specific antibody. We used uninfected cells and mock-infected cells (the vehicle) as controls. The cells were treated with a concentration of Shield-1 ranging from 10 to 1000 nM for 7 d. As shown, the treatment with Shield-1 was able to regulate the expression of DD-Cas9 in a strong dose-dependent manner (Figure 1B and Figure 2A). RT-PCR analysis with specific primers for DD-Cas9 and control primers for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) confirmed that levels of mRNA expression of DD-Cas9 were similar amongst the transduced cells and the vehicle notwithstanding the presence or absence of Shield-1 (Figure 2B). This confirms that the induction of Cas9 protein is a post-transcriptional event.
This system enables fast and reversible stabilization of the DD-Cas9 protein. Figure 3 shows sufficient induction of Cas9 expression in the transduced A549 cell line 2 h after treatment with 200 nM of Shield-1 compared to the uninfected A549 cells. However, withdrawal of Shield-1 results in a rapid decrease of the DD-Cas9 protein, which becomes negligible within 6 to 12 h (Figure 3).
The RPA3 protein is a component of the human replication protein A (RPA) heterotrimer. It is a single-stranded DNA binding complex that plays an important role in DNA replication, recombination, and repair. To validate the use of the system to study essential genes, we targeted the RPA3 gene. To this end, we used two independent locus-specific single guide RNAs (guides 25 and 44) as well as Renilla as a control (guide 208). The A549 cells transduced with the DD-Cas9 lentiviral construct were treated with 200 nM of Shield-1 for 3 d. A decrease in cell number in Shield-1-treated transduced A549 cells containing the RPA3 guide RNA was apparent after 48 h of treatment, and no effect was observed on the cell number in the Renilla sample (Figure 4A). To validate the depletion of the RPA3 protein, we performed an immunoblotting analysis using an antibody against RPA3 3 d after Sheild-1 induction (Figure 4B). Furthermore, we confirmed gene editing inversion or indel mutations using Surveyor nuclease assays and DNA sequencing (Figure 4C).
The lentiviral vector construct we designed bears a unique feature: the regulation of DD-Cas9 protein stability is independent of its mRNA expression. This enables the generation of bicistronic systems to express another gene of interest under the same EF-1a promoter without being modulated by the destabilized DD-Cas9 (Figure 5A,5B). We also added a 2A self-cleaving peptide (P2A) between DD-Cas9 and mVenus, a modified fluorescent protein that can be used to trace infected cells (Figure 5A). As mVenus is placed after P2A, as shown by the results of the Western Blot analysis in Figure 5B, the expression of the mVenus protein was observed in the vehicle and A549 transduced cells independent of DD-Cas9 expression and Shield-1 treatment.
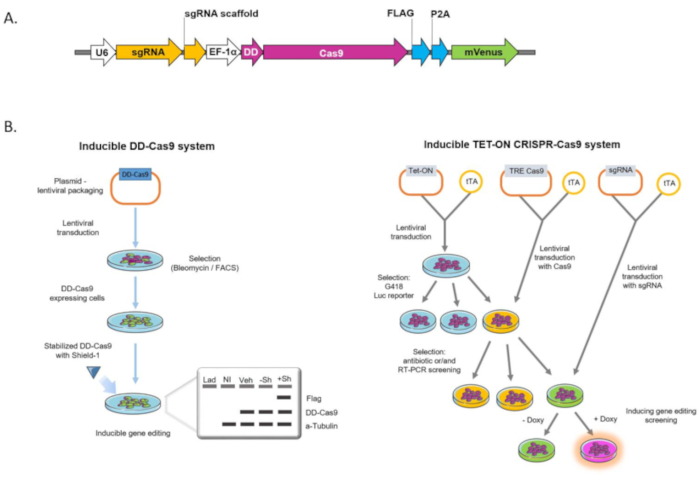
Figure 1: Schematic of the lentiviral construct and different gene-editing tools. A) The DD-Cas9 lentiviral backbone contains a U6 promoter, sgRNA, EF-1a promoter, DD, spCas9, nucleoplasmin NLS, and Flag-tag. B) Comparison between DD-Cas9 system (left panel) and different Tet-On system (right panel) used as gene-editing tools. Lad-ladder, NI-non-infected cells, Veh-vehicle, -Sh cells treated without Shield-1, + Sh cells treated with 200 nM Shield, – Doxy cells without doxycycline treatment, + Doxy cells treated with doxycycline. Please click here to view a larger version of this figure.
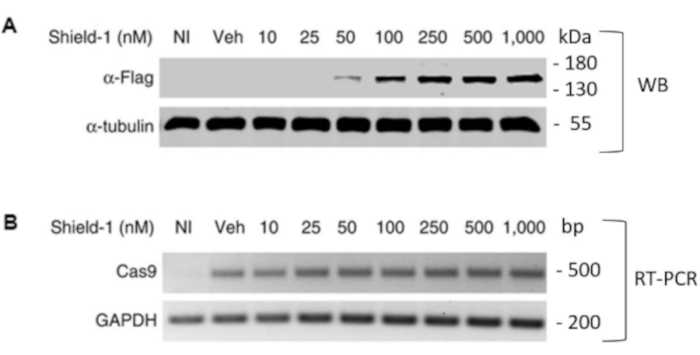
Figure 2: Representative results of dose-dependent DD-Cas9 stabilization by Shield-1. A) Western Blot analysis using an anti-Flag-tag antibody of stabilized DD-Cas9 expression in cells treated with increasing concentrations of Shield-1. As a control, the uninfected cells (NI) and mock-treated cells (Veh) were used. The fusion protein DD-Cas9 was undetectable in both controls. B) The RT-PCR results of mRNA expression levels of DD-Cas9 in transduced cells in the absence or dose-dependent treatments of Shield-1 were similar amongst transduced cells and vehicle, using GAPDH primers as an internal control. This figure has been modified from Serif et al19. Please click here to view a larger version of this figure.
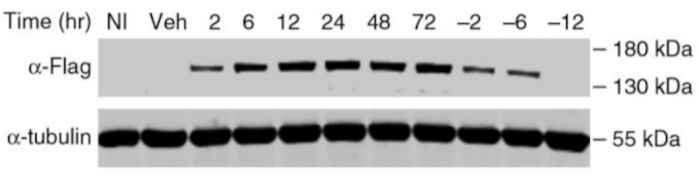
Figure 3: The Western Blot analysis illustrates the reversibility and rapidity of destabilized DD-Cas9 protein regulation after the withdrawal of its ligand Shield-1. Transduced A549 cell line with DD-Cas9 and uninfected A549 as control were treated 24 h after infection with 200 nM of Shield-1 ligand for the indicated time points. The protein level of DD-Cas9 in mock control cells was undetectable. This figure has been modified from Serif et al19. Please click here to view a larger version of this figure.
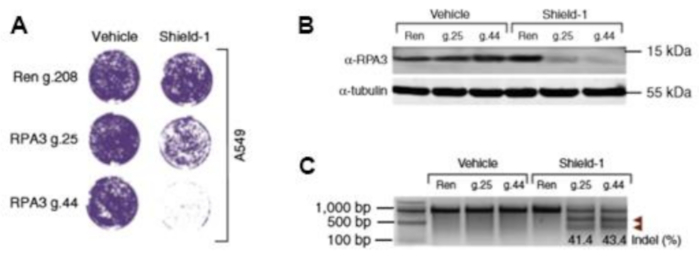
Figure 4: The DD-Cas9 system can induce robust gene editing in "in-vitro" and "in-vivo" settings. A) The cell line A549 transduced with a vector expressing sgRNA for RPA3 gene and DD-Cas9 (RPA3). As a control A549 transduced with a vector expressing sgRNA for Renilla (Ren) was used. Cells were treated with Shield-1 (200 nM) for 3 days which resulted in a rapid decrease in cell viability in cells expressing RPA3 sgRNA, with no effect on cell number in the Renilla control sample. The efficiency of RPA3 gene editing in the A549 cell line was validated by B) Western Blot analysis using the antibody against Flag-tag in RPA3 A549 and Renilla A549 in the presence and absence of 3 days Shield-1 (200 nM) treatment and C) by SURVEYOR nuclease assay. The arrows in panel C) show the fragments of the SURVEYOR nuclease assay. This figure has been modified from Serif et al19. Please click here to view a larger version of this figure.
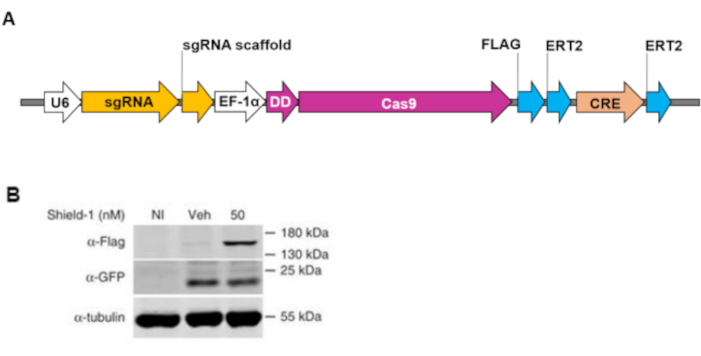
Figure 5: Scheme of a bicistronic DD-Cas9 lentiviral construct to drive the expression of mVenus independently of DD-Cas9. A) The construct consists of U6 promoter, sgRNA, EF-1a promoter, DD-Cas9, P2A, and mVenus. B) Transduced A549 cells with a lentiviral plasmid containing DD-Cas9, P2A, and mVenus were treated with 50 mM ligand Shield-1 for 3 days. On the third day, the western blot analysis was performed with cell lysate, using the antibody against GFP and Flag-tag separately. Figure 5B has been modified from Serif et al19. Please click here to view a larger version of this figure.
| Forward (Oligo 1) | 5’-CACCGNNNNNNNNNNNNNNNNNNNN-3’ |
| Reverse (Oligo 2) | 5’-AAACNNNNNNNNNNNNNNNNNNNNC-3’ |
Table 1:The design of the forward and reverse sgRNA oligonucleotide. The forward (Oligo 1) and the reverse (Oligo 2) oligonucleotide is designed by adding BsmBI enzyme digestion overhangs the your sgRNA sequence. “N” denotes the different nucleotides present in your sgRNA sequence, the rest are overhangs for BsmBI digestion.
| U6 primer sequence | 5’-GACTATCATATGCTTACCGT-3’ |
Table 2: The U6 promoter sequence for sgRNA cloning validation. To validate how successful is the sgRNA cloning, use the U6 promoter sequence for DNA sequencing of the DD-Cas9 plasmid.
