Neisseria meningitidis Infection of Induced Pluripotent Stem-Cell Derived Brain Endothelial Cells
Summary
The protocol described here highlights the major steps in the differentiating induced pluripotent stem-cell derived brain-like endothelial cells, the preparation of Neisseria meningitidis for infection, and sample collection for other molecular analyses.
Abstract
Meningococcal meningitis is a life-threatening infection that occurs when Neisseria meningitidis (meningococcus, Nm) can gain access to the central nervous system (CNS) by penetrating highly specialized brain endothelial cells (BECs). As Nm is a human-specific pathogen, the lack of robust in vivo model systems makes study of the host-pathogen interactions between Nm and BECs challenging and establishes a need for a human based model that mimics native BECs. BECs possess tighter barrier properties when compared to peripheral endothelial cells characterized by complex tight junctions and elevated trans-endothelial electrical resistance (TEER). However, many in vitro models, such as primary BECs and immortalized BECs, either lack or rapidly lose their barrier properties after removal from the native neural microenvironment. Recent advances in human stem-cell technologies have developed methods for deriving brain-like endothelial cells from induced pluripotent stem-cells (iPSCs) that better phenocopy BECs when compared to other in vitro human models. The use of iPSC-derived BECs (iPSC-BECs) to model Nm-BEC interaction has the benefit of using human cells that possess BEC barrier properties, and can be used to examine barrier destruction, innate immune activation, and bacterial interaction. Here we demonstrate how to derive iPSC-BECs from iPSCs in addition to bacterial preparation, infection, and sample collection for analysis.
Introduction
The blood-brain barrier (BBB), and the meningeal blood-CSF barrier (mBCSFB) are extremely tight cellular barriers that separate the circulation from the central nervous system (CNS) and are primarily comprised of highly specialized brain endothelial cells (BECs)1,2. Together, BECs maintain proper brain homeostasis by regulating nutrients and waste products in and out of the brain, while excluding many toxins, drugs, and pathogens1,2. Bacterial meningitis occurs when blood-borne bacteria are able to interact with, and penetrate the barrier formed by BECs and cause inflammation. Neisseria meningitidis (Nm, meningococcus) is a Gram-negative bacterium that colonizes the nasopharaynx of 10‒40 % of healthy individuals, but in some cases can cause serious systemic disease3. In affected individuals, Nm can gain access to the blood stream where it can cause purpura fulminans as well as penetrate BECs gaining access to the CNS causing meningitis3. Nm is a leading cause of bacterial meningitis world-wide, and despite vaccination efforts, is still a primary cause of meningitis4. Modern medical intervention, such as antibiotic treatment, have made these conditions survivable, however those affected with meningitis often are left with permanent neurological damage5,6.
Previous studies have identified bacterial factors and host signaling that contribute to Nm-BEC interactions7,8,9,10,11. The identified adhesins and invasins such as the opacity protein Opc, and type-IV pili, as well as receptors such as CD147, have been conducted on various BEC models in vitro, however these models lack many defining BBB properties7,9,11,12. Complete understanding of Nm-BEC interactions remain elusive due partially to the inability to utilize in vivo models, incomplete vaccination protection, and lack of robust human BEC models in vitro.
Modeling hBECs in vitro has been challenging due to the unique properties of BECs. Compared with peripheral endothelial cells, BECs have a number of phenotypes that enhance their barrier properties such as high trans-endothelial electrical resistance (TEER) due to complex tight junctions12. Once removed from the brain microenvironment, BECs rapidly lose their barrier properties limiting the usefulness of primary or immortalized in vitro models that only form a weak barrier12,13. The combination of the human specificity of Nm infections, lack of robust in vivo models, and challenges modeling human BECs in vitro creates a need for better models to understand the complex host-pathogen interaction between Nm and BECs. Recently using model human induced pluripotent stem cell (iPSC) technologies BEC-like cells have been derived from iPSCs that better mimic BECs in vivo12,13,14,15. iPSC-BECs are of human origin, easily scalable, and possess expected BEC phenotypes compared to their primary or immortalized counterparts12,13,14,15. Additionally we and others have demonstrated that iPSC-BECs are useful for modeling various diseases of the CNS such as host-pathogen interaction, Huntington’s disease, and MCT8 deficiency that causes Allan-Hurndon-Dudley syndrome16,17,18,19,20,21. Here, we demonstrate how to derive iPSC-BECs from renewable iPSC sources and the infection of iPSC-BECs with Nm leading to activation of the innate immune response. We believe that this model is useful to interrogate host-pathogen interaction that is unable to be recapitulated in other in vitro models and is especially useful when examining interactions with human specific pathogens such as Nm.
Protocol
NOTE: All media / reagent preparation, stem-cell maintenance, and differentiation steps are adapted from Stebbins et al.22.
1. Preparation of materials required for iPSC culture and BEC differentiation.
- Matrix coating of tissue culture (TC) plastic for IMR90-4 iPSC culture
- Aliquot basement membrane matrix gel (e.g., Matrigel) into 2.5 mg aliquots and store at -20 °C.
NOTE: Work quickly when handling the matrix gel and aliquot on ice, as it forms a gel above 4 °C and cannot be aliquoted once it has solidified. - For coating TC plastic, quickly add one aliquot of matrix gel to 30 mL of Dulbecco's Modified Eagle Medium (DMEM)/F12 medium in a 50 mL conical tube. Add 1 mL of the medium onto the frozen matrix gel, pipette up and down until it is thawed, and transfer immediately to the 50 mL conical tube with the remaining medium.
- Use 1 mL of this matrix gel-coating solution per well of a 6-well plate and 12 mL per T75 flask.
NOTE: Matrix gel-coated TC plastic can be prepared up to two weeks before it is used. It is, however, critical to avoid that the matrix gel solution dries out, which may require occasional addition of more DMEM/F12 on top of the wells.
- Aliquot basement membrane matrix gel (e.g., Matrigel) into 2.5 mg aliquots and store at -20 °C.
- Prepare stem-cell maintenance medium by adding 50 mL of 50x supplement to 450 mL of stem cell maintenance basal medium in the sterile environment of a biosafety cabinet.
NOTE: Other stem-cell maintenance media (mTeSR and E8) have been used in other studies13,14,15, 22,23,24,25. - To prepare 500 mL of unconditioned medium (UM), combine 392.5 mL of DMEM/F12 with 100 mL of knock out serum replacement (KOSR), 5 mL of non-essential amino acids, L-glutamine at a final concentration of 1 mM, and 3.5 µL of β-mercaptoethanol. Filter-sterilize and store at 4 °C for up to 2 weeks.
- To prepare 200 mL of endothelial cell (EC) medium plus retinoic acid (RA) plus bFGF, combine 198 mL of human endothelial serum free medium (hESFM), 2 mL of filter-sterilized platelet-poor derived serum (PDS), and 20 ng/mL bFGF. Filter sterilize and store for up to 2 weeks at 4 °C. Just before addition to cells, add 10 μM of RA to EC medium.
NOTE: As PDS has been discontinued and may therefore be limited, this protocol has been successfully conducted using B27 in place of PDS15,23,26. - To prepare 200 mL of EC medium without RA or bFGF, combine 198 mL of hESFM and 2 mL of filter sterilized PDS and filter sterilize. Store for up to 4 weeks at 4 °C.
2. Maintenance IMR90-4 cell culture
NOTE: Here we use the IMR90-4 cell line as an example, however other induced pluripotent stem-cell lines such as CC3, CD10, CD12, DF19-9-11T, 83iCTR, 00iCTR, and CS03iCTRn2 have been successfully employed for differentiation into BECs13,14,15,16,17,23,27,28.
- Culture iPSCs at 37 °C in 5% CO2 and typically maintain on 6-well plates at various densities with 2 mL of stem-cell maintenance medium per well.
- For maintenance of the iPSC culture, select a single well for passage that is not confluent and has open spaces between colonies.
- Aspirate the culture medium, add 1 mL of non-enzymatic cell dissociation reagent, and incubate at 37 °C for 7 min. While incubation is ongoing, replace the matrix gel solution, on a new 6-well plate, with 2 mL of fresh stem-cell maintenance medium per well.
- Carefully aspirate the non-enzymatic cell dissociation reagent taking care to not aspirate cells still attached to the plate.
- Add 6 mL of stem-cell maintenance medium and rinse the well bottom a few times until all cells are completely detached. Then, seed the new 6-well plate with varying densities, typically 1:6 or 1:12 for normal maintenance.
NOTE: Using the aforementioned ratios, the cells are split approximately twice a week. - Move the plate to the incubator and distribute the seeded cells equally across the wells by shaking the plate back and forth and left to right, pausing in between alternating shaking motions until the medium has settled.
3. Differentiation of brain endothelial cells from human iPSCs
- Plating of iPSCs for differentiation (day -3 of the differentiation protocol)
- Per IMR90-4 maintenance culture well used to plate for differentiation, aspirate the culture medium, add 1 mL of enzymatic cell dissociation reagent per well, and incubate at 37 °C for 7 min.
- Inactivate the enzymatic cell dissociation reagent by transferring the 1 mL of dissociated cell suspension into a 15 mL conical tube with at least 2 mL of fresh stem-cell maintenance medium per 1 mL of cells. Spin down the cell suspension at 1,500 x g for 5 min.
- Resuspend the cell pellet in 1 mL of stem-cell maintenance medium per well of IMR90-4 cells used, and count the cells using a hemocytometer.
NOTE: It may be helpful to dilute 1:1 with 0.4% trypan blue to distinguish between live and dead cells when counting. Depending on the density of iPSCs, one well of a 6-well plate usually yields 1‒2 x 106 cells. - For differentiation in a T75 flask, add 7.5 x 105 cells to 12 mL of stem-cell maintenance medium and ROCK inhibitor (Y27632 dihydrochloride) at a final concentration of 10 µM. Aspirate matrix gel-coating solution from a T75 flask and transfer the cell suspension to the flask. Distribute the cells equally by shaking the flask back and forth and left to right (see step 2.2.4) and incubate at 37 °C and 5% CO2.
NOTE: It is critical to add ROCK inhibitor at this step to enhance survival of the dissociated single stem cells25,29. Cells should be evenly distributed across the flask and in singlets exhibiting a spread, mesenchymal-like morphology due to the ROCK inhibitor treatment22.
- On day -2 and day -1, change media to fresh stem-cell maintenance medium; 12 mL per T75.
- On day 0, start differentiation by changing media to UM; 12 mL per T75.
- Change UM daily (day 1 – day 5).
NOTE: The cells typically reach confluence after 2 to 3 days in UM, which can be observed with the naked eye or through an inverted bright field microscope. As the differentiation progresses, nestin+ “neural tracts” become visible with PECAM-1+ cells in between as previously described13,14. - Selectively expand the endothelial cell population by switching to EC medium with 20 ng/mL bFGF and 10 µM retinoic acid (RA) (day 6) and incubating for two days. EC medium with bFGF can be prepared up to two weeks prior. RA is added from frozen stock on the day of use (e.g., 1 µL of 10 mM RA stock per 1 mL of EC + bFGF).
NOTE: Successful differentiation can also be achieved without supplementation of RA at days 6 and 8. Omission of RA will, however, yield BECs with reduced TEER12,13,14. - Coat cell culture plates and membrane inserts (e.g., Transwell) with collagen IV and fibronectin (day 7), for purification of the BECs and following experiments.
- For coating of membrane inserts, combine 4 parts collagen IV (1 mg/mL in 0.5 mg/mL acetic acid), 1 part fibronectin (1 mg/mL) and 5 parts sterile tissue-grade water. ECM solution can be diluted 1:5 for coating cell culture plates (i.e., 4 parts collagen IV, 1 part fibronectin, 45 parts water). Incubate with coating solution at 37 °C overnight.
NOTE: Plates and membrane inserts can also be coated for at least 4 h prior to subculturing on the same day of the purification step.
- For coating of membrane inserts, combine 4 parts collagen IV (1 mg/mL in 0.5 mg/mL acetic acid), 1 part fibronectin (1 mg/mL) and 5 parts sterile tissue-grade water. ECM solution can be diluted 1:5 for coating cell culture plates (i.e., 4 parts collagen IV, 1 part fibronectin, 45 parts water). Incubate with coating solution at 37 °C overnight.
- Purify BECs by subculturing the differentiated cells on collagen IV and fibronectin-coated plates or membrane inserts (day 8).
- Aspirate EC medium and add enzymatic cell dissociation reagent (12 mL per T75). Incubate at 37 °C until 90% of cells have detached from the flask.
NOTE: Cell dissociation can take up to 1 h. - During the incubation time, remove the collagen IV/fibronectin coating solution from previously prepared plates/inserts and let them dry in a sterile hood. It takes approximately 20 min for the inserts to dry.
- Once the cells have detached, wash them off the flask using a 10 mL pipette. Pipette up and down to achieve a single cell suspension.
NOTE: Single cell suspension is important for reliable cell counting and to achieve solid monolayers. - Dilute with at least equal volume of fresh hESFM in a 50 mL conical tube and count cells using a hemocytometer.
- Pellet the cells at 1,500 x g for 10 min.
- Resuspend cells in appropriate volume of freshly prepared EC + bFGF + RA to achieve a suspension of 2 x 106 cells/mL for seeding on membrane inserts. Add 500 µL (1 x 106 cells) on top of a 12-well insert and 1.5 mL of EC + bFGF + RA medium on the bottom. For seeding on 24 and 48-well plates, dilute cell suspension 1:2 and add 500 µL (5 x 105 cells) and 250 µL (2.5 x 105 cells) per well, respectively. Distribute cells evenly across the well/insert (see step 2.2.4) and incubate at 37 °C under 5% CO2.
- Aspirate EC medium and add enzymatic cell dissociation reagent (12 mL per T75). Incubate at 37 °C until 90% of cells have detached from the flask.
- Change media on plates/transwells to EC without bFGF or RA (day 9).
- Conduct infection experiments, TEER measurement, and immunofluorescence staining as described in the following sections (day 10).
NOTE: Successfully differentiated and purified BECs typically reach peak TEER on day 10 and express characteristic markers of brain endothelial cells such as PECAM-1 (CD31) and VE-cadherin, the glucose transporter GLUT-1, efflux transporters such as p-glycoprotein, and tight junction components ZO-1, Occludin, and Claudin-5 13,14,16,17,19,22. Refer to Lippmann et al., Stebbins et al. and others for further details and images of the cell types, morphologies, and expression of cell type specific markers during the differentiation process13,14,15,16,17,19,22. Representative images of the IMR90-4 cells at different stages in the BEC differentiation process can be found in Supplementary Figure 1.
4. Transendothelial electrical resistance (TEER) as a measure of barrier tightness
NOTE: TEER is usually read on membrane inserts on days 9 and 10 of differentiation to confirm successful generation of barrier forming iPSC-BECs (Figure 1A).
- Place the epithelial volt-ohm meter (EVOM) in the sterile environment of a biosafety hood and connect the electrode to the EVOM.
- Disinfect the electrode by submerging it in 70% EtOH for at least 5 min and let it dry completely.
NOTE: Longer incubation in 70% EtOH or decontamination of the electrode using 5% hypochlorite solution is possible if needed. - Retrieve the iPSC-BECs on membrane inserts from the incubator and measure TEER.
NOTE: It is important to read TEER rapidly after removal from the incubator as temperature change may impact TEER measurement. - Read TEER by dipping the electrode into the medium so that the shorter electrode is placed on top of the insert and the longer electrode reaches into the medium surrounding the insert.
NOTE: Make sure the electrodes at the tips of the “chopsticks” are completely covered by liquid. If needed, tilt the well to achieve this before setting the plate down again for measuring.
5. Immunofluorescence (IF) staining to validate BEC phenotype
NOTE: To validate the quality of the fully differentiated and purified cells, iPSC-BEC monolayers are stained for the characteristic markers of brain endothelial cells on day 10 of the differentiation process as previously described (Figure 1B‒G) 13,14,15,16,17,19,22.
- Aspirate the medium and wash 1x with PBS.
- Fix cells with ice cold methanol at room temperature (RT) for 15 min.
- Wash 3x with phosphate buffered saline (PBS) and block with 10% fetal bovine serum (FBS) in PBS at RT for 1 h.
- Aspirate and add primary antibodies diluted in blocking solution. Incubate at 4 °C overnight.
NOTE: Refer to the Table of Materials and Stebbins et al. for information related to source and dilution of the antibodies22. - Wash 3x with PBS before adding secondary antibody diluted in blocking solution. Incubate at RT for 1 h. Protect samples from light from this point on.
- Wash 2x with PBS. Then, add DAPI at a 1:5,000 dilution in PBS and stain at RT for 15 min.
- Wash 1x leaving the PBS on and take images using a fluorescence microscope.
6. Preparation of bacteria and infection of iPSC-BECs
- On the day before the infection experiment (day 9 of differentiation), start an overnight culture of the bacteria from frozen stock. For infection with Nm, streak bacteria onto Columbia agar with 5% sheep blood (blood-agar). Incubate bacterial cultures at 37 °C and 5% CO2 overnight.
- The next day (day 10), prepare fresh PPM+ by supplementing Protease peptone media (PPM) with 50 µL of 2 M MgCl2, 50 µL of 2 M NaHCO3, and 100 µL Kellogg’s supplement per 10 mL. Inoculate 10 mL of PPM+ medium in a 50 mL conical tube with Nm from the overnight culture plate using a sterile cotton swab. Incubate shaking at 200 rpm at 37 °C for 1.5 h (i.e., until bacteria are in logarithmic growth phase).
- During bacterial incubation, prepare iPSC-BECs in a 24-well plate or on membrane inserts for infection by replacing the old medium with 400 µL of fresh EC medium (without bFGF or RA) per well/ top of membrane insert.
- Centrifuge bacterial culture at 4000 x g for 10 min and aspirate the media. Resuspend the bacterial pellet in 250 µL of PBS.
- In 4 mL of fresh PBS, use a portion of the bacterial cell suspension and adjust to an OD600 of 0.4 (approximately 1 x 108 CFU/mL).
- Then, dilute the bacteria in cell culture medium (EC medium) according to the desired multiplicity of infection (MOI).
NOTE: For example, for an MOI of 10, dilute 1:10 or 1:5 when infecting iPSC-BECs in a 24-well plate or on membrane inserts, respectively (1 x 105 cells per monolayer in a 24-well plate). Addition of human serum is not included in the preparation of Nm for infection as was described in other manuscripts as it was observed to have a deleterious impact on the iPSC-BEC barrier phenotype as measured by TEER (data not shown)30,31. However, the interaction of Nm and iPSC-BECs is unaffected with or without human serum (data not shown). - Infect iPSC-BECs in each well with 100 µL of the prepared bacteria suspension and incubate for the desired time of infection.
NOTE: Expression of a number of proinflammatory cytokines and chemokines is elevated in iPSC-BECs when infected with Nm, most prominently after 8 h of infection as previously described by Martins-Gomes et al (Figure 2)19.
7. Innate immune activation by quantitative PCR
NOTE: Using a preferred RNA isolation, cDNA synthesis, and qPCR protocol, collect samples and run qPCR on selected cytokines.
- 7.1. Collect the RNA from BEC samples after infection on day 10 of the differentiation protocol, using reagents form a commercially available RNA isolation kit (see the Table of Materials).
NOTE: Avoid nuclease contamination of samples by working carefully in a cleaned and sterilized environment.- Prepare lysis buffer and add 350 µL to each well/monolayer of iPSC-BECs.
- Collect the samples by pipetting up and down numerous times (e.g., 20x) and transferring the suspension to a sterile microcentrifuge tube.
NOTE: Samples can be stored at -80 °C until ready for RNA isolation. - Follow the protocol provided with the RNA isolation kit for RNA purification from cultured cells and tissue.
- After elution in nuclease-free water, keep RNA samples on ice to minimize any potential RNase activity.
- Estimate RNA concentrations on a spectrophotometer (e.g., Nanodrop).
- Generate a cDNA library from the collected RNA using a cDNA synthesis kit (see Table of Materials).
- Set up reactions consisting of cDNA synthesis master mix, at least 200 ng (ideally 500 ng) of sample RNA, and nuclease-free water in a defined total reaction volume as described in the protocol of the cDNA synthesis kit.
- On a standard thermo cycler, run a program that is appropriate for the reagents of the cDNA synthesis kit used. For instance: 25 °C for 2 min, 55 °C for 10 min, 95 °C for 1 min.
- After synthesis, dilute the cDNA up to 1:10 in nuclease-free water and move samples to 4 °C for short term or -20 °C for long term storage.
NOTE: Lower dilution may be necessary if RNA concentration was low (e.g., below 50 ng). The diluted cDNA can be stored at -20 °C for up to a year.
- Perform qPCR on the cDNA samples targeting transcripts of innate immune response genes such as cytokines and chemokines with carefully designed and validated primers.
NOTE: As primer design is very important for the quality of qPCR results, primer efficiency should be tested conducting a dilution series and the absence of multiple products should be confirmed on a DNA gel.- Per 25 µL reaction, use 0.5 µL forward and 0.5 µL reverse primer (10 mM in nuclease-free water), 1 µL cDNA, 10.5 µL nuclease free H2O, and 12.5 µL qPCR master mix.
- Perform the reaction on a qPCR machine using the following cycler protocol: (a) 4 °C for 2 min; (b) 95 °C for 15 min; (c) 95 °C for 15 s; (d) 60 °C for 1 min; cycle through (c) and (d) 45 times; (e) optional melt curve: 30‒99 °C in 1 °C increments; 25 °C for 5 min.
- For data analysis, use the ΔΔCT calculation to compare cytokine and chemokine expression levels to a reference housekeeping gene such as 18S or GAPDH.
Representative Results
The protocol described here is adapted from Stebbins et al. and highlights the process to differentiate iPSCs into brain-like endothelial cells that possess BBB properties, and how to utilize this model for infection studies using iPSC-BECs with Nm19,22. The iPSC-BECs, when differentiated properly, exhibit tight barrier properties measured by TEER that are often greater than 2000 Ω·cm2, and express endothelial markers such as VE-cadherin and CD31 (PECAM) (Figure 1A‒C). Additionally, they express and localize the tight junction markers Claudin-5, Occludin, and ZO-1 (Figure 1D‒F), and transporters such as Glut-1 (Figure 1G). Upon infection with Nm, iPSC-BECs respond to infection through the upregulation of neutrophilic proinflammatory cytokines as measured by qPCR such as IL-8 (CXCL8), CXCL1, CXCL2, CCL20, and IL6 (Figure 2A‒E). These representative results demonstrate how to ensure that iPSC-BECs are being differentiated reliably, and how to examine the response of iPSC-BECs to Nm infection.
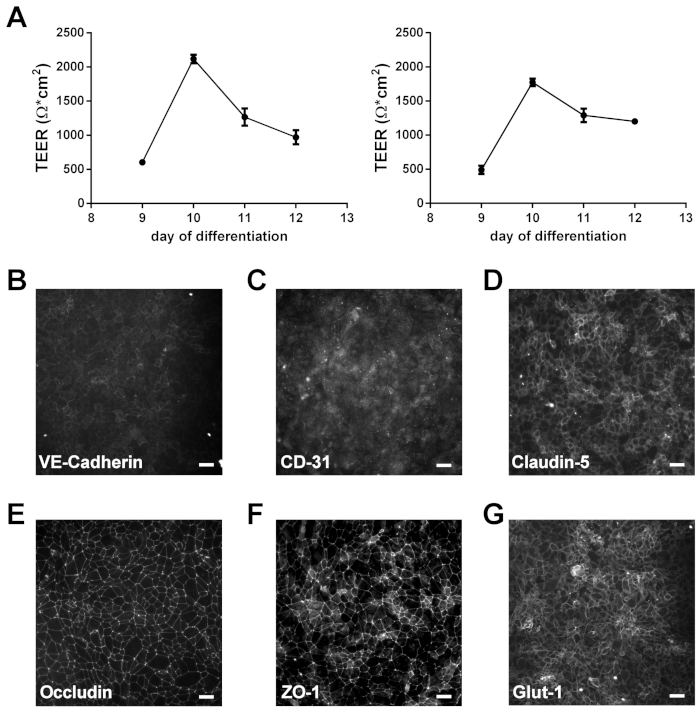
Figure 1: Characterization of iPSC-BECs. (A) TEER of two separate, individual differentiations, read on days 9–12. Data presented as mean of triplicates. Error bars represent ± SD. (B‒G) Representative immunofluorescence data showing expression of endothelial cell markers VE-cadherin (B) and PECAM-1 (CD31; C), tight junction components Claudin-5 (D), Occludin (E), and ZO-1 (F), and glucose transporter GLUT-1 (G). Scale bar represents 50 μm. Panels B‒G of this figure have been used with permission from Kim et al. originally published in Fluids and Barriers of the CNS, a BMC journal17. Please click here to view a larger version of this figure.
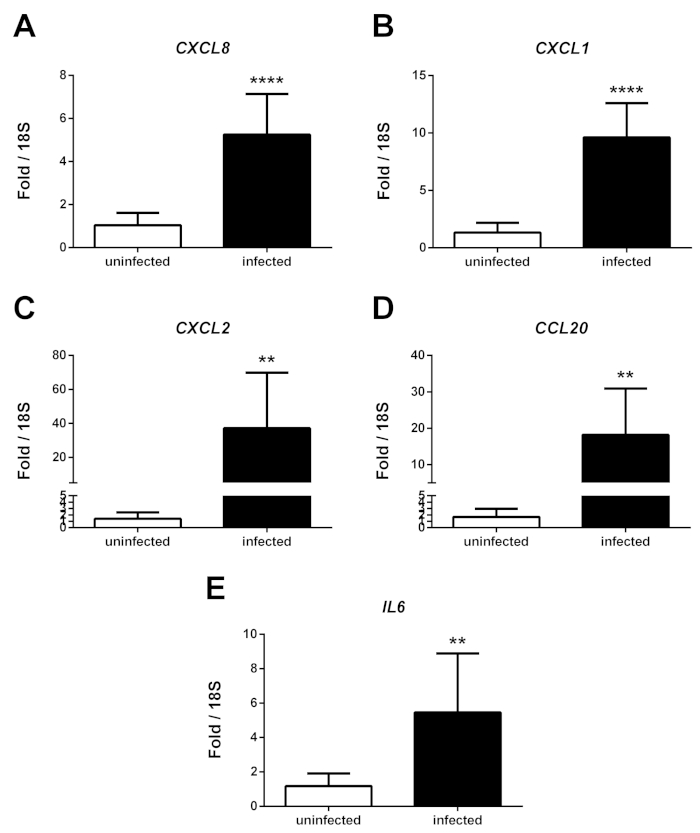
Figure 2: Upregulation of cytokines by iPSC-BECs upon infection with Neisseria meningitidis. Representative qPCR data showing relative expression of CXCL8 (A), CXCL1 (B), CXCL2 (C), CCL20 (D), and IL6 (E) transcripts after 8 h of infection, comparing infected with uninfected iPSC-BEC monolayers. Data presented as mean of three independent experiments conducted in triplicate. Error bars represent ± S.D. Student’s t-test used to determine significance. *p < 0.05; **p < 0.01; ***p < 0.001. This figure has been modified and used with permission from Martins Gomes et al. originally published in Frontiers in Microbiology19. Please click here to view a larger version of this figure.
Supplementary Figure 1: IMR90-4 cells at different stages in the BEC differentiation process. Representative images of maintenance IMR90-4 culture ready to be passaged (A) and cells at different stages in the BEC differentiation process: (B) after seeding with ROCK inhibitor (day -2), (C) at the start of differentiation (day 0), (D) first day of confluence (day 3), (E) end of UM phase (day 6), (F) before BEC purification (day 8), and (G and H) after BEC purification (day 9 and day 10). Scale bar represents 500 µm. Please click here to view a larger version of this figure.
Discussion
Modeling BECs and the BBB has had challenges, as primary and immortalized human BECs, in vitro, tend to lack robust barrier phenotypes. The advent of human stem cell technologies has allowed for the generation of iPSC derived BEC-like cells that retain expected hallmark BBB phenotypes such as endothelial markers, tight junction expression, barrier properties, response to other CNS cell types, and functional efflux transporters12,13,14,15, 22, 24, 25. This has enabled researchers to utilize BECs in vitro that closely mimic in vivo BECs and model various diseases with reported BBB dysfunction16,17,19,20,21,32. Nm is a leading cause of bacterial meningitis and is a human specific pathogen that lacks robust in vivo models19. This limitation has necessitated the use of better engineered models to drive the discovery of novel host-pathogen interaction between Nm and the BBB. Recently, we have demonstrated that iPSC-BECs are a viable model to interrogate Nm-BEC interaction19.
Here we describe a general method to derive iPSC-BECs and infect with Nm resulting in the upregulation of proinflammatory cytokines that are typically induced by bacterial infection19. For the derivation of iPSC-BECs we generally follow the protocol as described in Stebbins et al. for the generation of iPSC-BECs, with minor modifications22. In particular here we use StemFlex media instead of mTesR1, however either media can be used for the maintenance of the stem cell culture17. It has been established that this protocol works well with many iPSC lines, however it is important that the optimum seeding density is determined for each individual iPSC line15, 24. For this manuscript we used the IMR90-4 cell line and it was previously established that 1 x 105 cells/cm2 was the optimum initial seeding density24. Finally as a demonstration of the identity of BECs generated, iPSC-BECs express expected endothelial cell markers and tight junctions while exhibiting high TEER (Figure 1)13,14,15,24. These phenotypes, as well as being of human origin, make iPSC-BECs a powerful tool to interrogate Nm-BEC interaction.
The preparation of Nm for infection was adapted from previously published methods19,33. To ensure that the bacterial growth media was not introduced into the cell culture experiments, a washing step and resuspension in PBS was conducted as described in the methods. Finally, an MOI of 10 had been previously observed to result in the activation of iPSC-BECs through an upregulation of pro-inflammatory cytokines19. Activation of BECs in response to various bacteria have been observed namely through the upregulation of neutrophilic chemokines and cytokines6. It has been previously observed that iPSC-BECs upregulate the potent neutrophil chemoattractants IL-8, CXCL1, and CXCL2 after infection with Group B Streptococcus, and Nm16,19. This observed response of iPSC-BECs demonstrate that these cells are able to detect bacteria and activate an innate immune program resulting in the upregulation of cytokines. The methods to detect the upregulation of these cytokines by qPCR are well established and are briefly described above. However interestingly, while these pro-inflammatory cytokines are detected by qPCR, the coordinate protein products are either undetected or very low16,19. At present, it is unclear if this is an artifact of the iPSC-BEC models, or if the observed low abundance of cytokines is biologically relevant. Future research will be required to determine a mechanism behind the disconnect between expression and secretion.
A major strength of the iPSC-BEC model is the expression and localization of tight junctions that contribute to barrier function as read be TEER12,13,14,15,22. Previous work with Streptococcus agalactiae (Group B Streptococcus, GBS) has demonstrated that the upregulation of Snail1 contributes to the destruction of BBB tight junctions in vitro and in vivo34. More recently, this finding was confirmed in the iPSC-BEC model both with GBS and Nm suggesting a mechanism for how bacteria are able to disrupt BBB integrity during infection16,19. Additionally, it was demonstrated that Nm interacts with CD147 on endothelial cells that promote bacterial attachment, and ultimately reorganization of tight junctions leading to barrier dysfunction9. We have demonstrated that Nm colocalizes with CD147 in the iPSC-BECs potentially making this model ideal for the future elucidation of Nm-CD147 interactions as they pertain to BBB dysfunction19.
The method presented here demonstrates the differentiation of iPSC-BECs from a pluripotent stem cell source, and application with Nm infection. The iPSC-BECs are of human origin, express endothelial markers, and possess BBB specific phenotypes making them an ideal model for the examination of human specific pathogens such as Nm. Finally, we are able to demonstrate that the iPSC-BEC model respond to bacterial infection through the upregulation of a neutrophilic cytokine response. Taken together, the iPSC-BEC model has certain advantages over primary and immortalized model systems to examine the host-pathogen interactions at the BBB. Further work should be aimed at elucidating mechanisms of BBB destruction during bacterial meningitis.
Disclosures
The authors have nothing to disclose.
Acknowledgements
L.M.E. is supported by the DFG research training program GRK2157 entitled “3D Tissue Models for Studying Microbial Infections by Human Pathogens” awarded to A. S-U. B.J.K. is supported by a postdoctoral fellowship by the Alexander von Humboldt Foundation. Additionally, we acknowledge Lena Wolter for her technical assistance in the generation of the iPSC-BECs in culture.
Materials
| Accutase (1x) | Sigma | A6964 | Enzymatic cell dissociation reagent |
| Acetic acid | Sigma | A6283 | |
| All-trans retinoic acid (RA) | Sigma | R2625 | |
| Anti-CD31 (PECAM-1) | Thermo Scientific (Labvision) | RB-10333 | |
| Anti-Claudin-5 | Invitrogen | 4C3C2 | |
| Anti-Glut-1 | Thermo Scientific (Labvision) | SPM498 (MA5-11315) | |
| Anti-Occludin | Invitrogen | 33-1500 | |
| Anti-VE-cadherin | Santa Cruz | sc-52751 | |
| Anti-ZO-1 | Invitrogen | 33-9100 | |
| Bacto Proteose Peptone | BD | 211684 | |
| b-Mercaptoethanol | Merck (Sigma-Aldrich) | 805740 | |
| Cell culture plates and flasks | Sarstedt | ||
| Centrifuge (Heraeus Megafuge 1.0R) | Thermo Scientific | ||
| Class II biosafety cabinet | Nuaire | NU-437-400E | |
| CO2 Incubator (DHD Autoflow CO2 Air-Jacketed Incubator) | Nuaire | ||
| Collagen IV | Sigma | C5533 | |
| Columbia ager + 5 % sheep blood | Biomerieux | 43049 | |
| Costar Transwell polyester filters (12- or 24-well) | Corning | 3460, 3470 | |
| D(+)-Glucose | Merck (Sigma-Aldrich) | G8270 | |
| DAPI | Invitrogen | D1306 | |
| DMEM/F12 | Gibco | 31330-038 | |
| DMSO | ROTH | A994.1 | |
| Dulbecco's phosphate-buffered saline (DPBS) | Gibco | 21600-069 | |
| Epithelial Volt-Ohm Meter (Millicell ERS-2) with STX electrode | Merck (Millipore) | MERS00002 | |
| Fe(NO3)3 | ROTH | 5632.1 | |
| Fibronectin | Sigma | F1141 | |
| Fluoresence microscope (Eclipse Ti) | Nikon | ||
| Hemacytometer (Neubauer) | A. Hartenstein | ZK06 | |
| Human basic fibroblast growth factor (bFGF) | PeproTech | 100-18B | |
| Human Endothelial Serum Free Medium (hESFM) | Gibco | 11111-044 | |
| Inverted microscope (Wilovert) | Hund (Will Wetzlar) | ||
| iPS(IMR90)-4 cells | WiCell | ||
| Kellogg's supplement | To prepare 110 ml of Kellogg's supplement, prepare 100 ml of 4 g/ml glucose, 0.1 g/ml glutamine, and 0.2 mg/ml thiamine pyrophosphate and 10 ml of 5 mg/ml Fe(NO3)3 and combine the solutions. Filter sterilize and store aliquoted at -20 °C. | ||
| Knockout serum replacement (KOSR) | Gibco | 10828-028 | |
| L-glutamine (GlutaMAX) | Invitrogen | 35050-038 | |
| LunaScript RT SuperMix Kit | NEB | E3010L | cDNA synthesis kit |
| Matrigel Matrix | Corning | 354230 | |
| Methanol | ROTH | 4627.5 | |
| MgCl2 | ROTH | KK36.1 | |
| Micropipettes (Research Plus) | Eppendorf | ||
| NaHCO3 | ROTH | 6329 | |
| Nonessential amino acids (NEAA) | Gibco | 11140-035 | |
| NucleoSpin RNA isolation kit | Machery-Nagel | 740955 | RNA isolation kit |
| Pipette boy (Accu-Jet Pro) | Brand | ||
| Platelet poor plasma-derived serum, bovine (PDS) | Fisher | 50-443-029 | |
| PowerUp SYBR Green Master Mix | Applied Biosystems | A25742 | qPCR master mix |
| qPCR film (MicroAmp Optical Adhesive Film) | Applied Biosystems | 4211971 | |
| qPCR plates (MicroAmp Fast 96-well) | Applied Biosystems | 4346907 | |
| ROCK inhibitor, Y27632 dihydrochloride | Tocris | 1254 | |
| RT-PCR thermo cycler (StepOnePlus) | Applied Biosystems | 4376600 | |
| Serological pipettes | Sarstedt | ||
| StemFlex basal medium + 50x StemFlex supplement | Gibco | A3349401 | Stem-cell maintenance medium |
| Swinging Bucket Rotor (Heraeus #2704) | Thermo Scientific | ||
| Thiamine pyrophosphate | Sigma | C8754-5G | |
| Trypan Blue Solution, 0.4% | Gibco | 15250061 | |
| Versene | Gibco | 15040-033 | Non-enzymatic cell dissociation reagent (EDTA) |
References
- Rua, R., McGavern, D. B. Advances in Meningeal Immunity. Trends in Molecular Medicine. 24 (6), 542-559 (2018).
- Abbott, N. J., Patabendige, A. A. K., Dolman, D. E. M., Yusof, S. R., Begley, D. J. Structure and function of the blood-brain barrier. Neurobiology of Disease. 37 (1), 13-25 (2010).
- Rouphael, N. G., Stephens, D. S. Neisseria meningitidis: Biology, microbiology, and epidemiology. Methods in Molecular Biology. , (2012).
- Stephens, D. S., Greenwood, B., Brandtzaeg, P. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet. 369 (9580), 2196-2210 (2007).
- Le Guennec, L., Coureuil, M., Nassif, X., Bourdoulous, S. Strategies used by bacterial pathogens to cross the blood-brain barrier. Cellular Microbiology. , (2020).
- Doran, K. S., et al. Host-pathogen interactions in bacterial meningitis. Acta Neuropathologica. 131 (2), 185-209 (2016).
- Cunha, C. S. E., Griffiths, N. J., Virji, M. Neisseria meningitidis opc invasin binds to the sulphated tyrosines of activated vitronectin to attach to and invade human brain endothelial cells. PLoS Pathogens. , (2010).
- Coureuil, M., et al. Meningococcus hijacks a β2-adrenoceptor/β-arrestin pathway to cross brain microvasculature endothelium. Cell. 143 (7), 1149-1160 (2010).
- Bernard, S. C., et al. Pathogenic Neisseria meningitidis utilizes CD147 for vascular colonization. Nature Medicine. 20 (7), 725-731 (2014).
- Slanina, H., Hebling, S., Hauck, C. R., Schubert-Unkmeir, A. Cell invasion by neisseria meningitidis requires a functional interplay between the focal adhesion kinase, Src and cortactin. PLoS ONE. 7 (6), (2012).
- Unkmeir, A., et al. Fibronectin mediates Opc-dependent internalization of Neisseria meningitidis in human brain microvascular endothelial cells. Molecular Microbiology. 46 (4), 933-946 (2002).
- Helms, H. C., et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. Journal of Cerebral Blood Flow and Metabolism. , (2015).
- Lippmann, E. S., et al. Derivation of Blood-Brain Barrier Endothelial Cells from Human Pluripotent Stem Cells. Nature Biotechnology. 30 (8), 783-791 (2012).
- Lippmann, E. S., Al-Ahmad, A., Azarin, S. M., Palecek, S. P., Shusta, E. V. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Scientific Reports. 4, 4160 (2014).
- Hollmann, E. K., Bailey, A. K., Potharazu, A. V., Neely, M. D., Bowman, A. B., Lippmann, E. S. Accelerated differentiation of human induced pluripotent stem cells to blood-brain barrier endothelial cells. Fluids and Barriers of the CNS. , (2017).
- Kim, B. J., et al. Modeling Group B Streptococcus and Blood-Brain Barrier Interaction by Using Induced Pluripotent Stem Cell-Derived Brain Endothelial Cells. mSphere. , (2017).
- Kim, B. J., et al. Streptococcus agalactiae disrupts P-glycoprotein function in brain endothelial cells. Fluids and Barriers of the CNS. 16 (1), 26 (2019).
- Kim, B. J., Shusta, E. V., Doran, K. S. Past and Current Perspectives in Modeling Bacteria and Blood–Brain Barrier Interactions. Frontiers in Microbiology. , (2019).
- Martins Gomes, S. F., et al. Induced Pluripotent Stem Cell-Derived Brain Endothelial Cells as a Cellular Model to Study Neisseria meningitidis Infection. Frontiers in Microbiology. , (2019).
- Vatine, G. D., et al. Modeling Psychomotor Retardation using iPSCs from MCT8-Deficient Patients Indicates a Prominent Role for the Blood-Brain Barrier. Cell Stem Cell. , (2016).
- Lim, R. G., et al. Huntington’s Disease iPSC-Derived Brain Microvascular Endothelial Cells Reveal WNT-Mediated Angiogenic and Blood-Brain Barrier Deficits. Cell Reports. 19 (7), 1365-1377 (2017).
- Stebbins, M. J., Wilson, H. K., Canfield, S. G., Qian, T., Palecek, S. P., Shusta, E. V. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods. 101, 93-102 (2016).
- Neal, E. H., et al. A Simplified, Fully Defined Differentiation Scheme for Producing Blood-Brain Barrier Endothelial Cells from Human iPSCs. Stem Cell Reports. 12 (6), 1380-1388 (2019).
- Wilson, H. K., Canfield, S. G., Hjortness, M. K., Palecek, S. P., Shusta, E. V. Exploring the effects of cell seeding density on the differentiation of human pluripotent stem cells to brain microvascular endothelial cells. Fluids and Barriers of the CNS. , (2015).
- Wilson, H. K., Faubion, M. G., Hjortness, M. K., Palecek, S. P., Shusta, E. V. Cryopreservation of brain endothelial cells derived from human induced pluripotent stem cells is enhanced by rho-associated coiled coil-containing kinase inhibition. Tissue Engineering – Part C: Methods. , (2016).
- Qian, T., et al. Directed differentiation of human pluripotent stem cells to blood-brain barrier endothelial cells. Science Advances. , (2017).
- Sances, S., et al. Human iPSC-Derived Endothelial Cells and Microengineered Organ-Chip Enhance Neuronal Development. Stem Cell Reports. , (2018).
- Canfield, S. G., et al. An isogenic blood-brain barrier model comprising brain endothelial cells, astrocytes, and neurons derived from human induced pluripotent stem cells. Journal of Neurochemistry. , (2017).
- Kurosawa, H. Application of Rho-associated protein kinase (ROCK) inhibitor to human pluripotent stem cells. Journal of Bioscience and Bioengineering. , (2012).
- Schubert-Unkmeir, A., Sokolova, O., Panzner, U., Eigenthaler, M., Frosch, M. Gene expression pattern in human brain endothelial cells in response to Neisseria meningitidis. Infection and Immunity. 75 (2), 899-914 (2007).
- Sokolova, O., et al. Interaction of Neisseria meningitidis with human brain microvascular endothelial cells: Role of MAP- and tyrosine kinases in invasion and inflammatory cytokine release. Cellular Microbiology. 6 (12), 1153-1166 (2004).
- Alimonti, J. B., et al. Zika virus crosses an in vitro human blood brain barrier model. Fluids and Barriers of the CNS. , (2018).
- Kim, B. J., Schubert-unkmeir, A. In vitro Models for Studying the Interaction of Neisseria meningitidis with Human Brain Endothelial Cells. Neisseria meningitidis: Methods and Protocols, Methods in Molecular Biology. 1969, 135-148 (2019).
- Kim, B. J., et al. Bacterial induction of Snail1 contributes to blood-brain barrier disruption. Journal of Clinical Investigation. 125 (6), 2473-2483 (2015).
