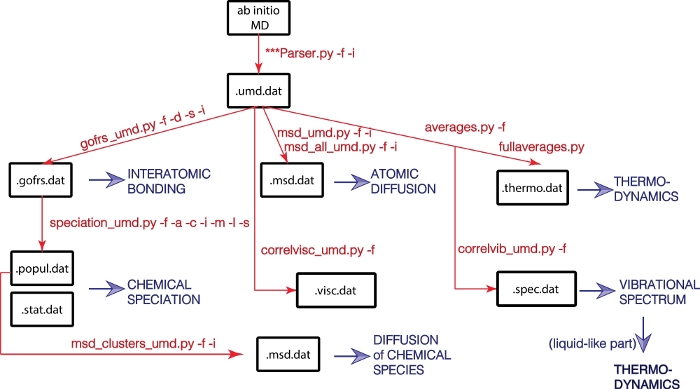
1. Analyse af molekylær-dynamik kører BEMÆRK: Pakken er tilgængelig via GitHubs hjemmeside (https://github.com/rcaracas/UMD_package) og via en dedikeret side (http://moonimpact.eu/umd-package/) af ERC IMPACT-projektet som en open-access-pakke. Uddrag hvert enkelt sæt fysiske egenskaber ved hjælp af et eller flere dedikerede Python-scripts fra pakken. Kør alle scripts på kommandolinjen. de anvender alle en række flag, som er så konsekvente som muligt fra et script til et andet. Flagene, deres betydning og standardværdierne er alle opsummeret i tabel 1. Flag Betydning Script ved hjælp af det Standardværdi -h Kort hjælp al -f UMD-filnavn al -I Termiske trin, der skal kasseres al 0 -I Inputfil, der indeholder de interatomare bindinger artsdannelse bonds.input -s Prøveudtagning af hyppigheden msd, speciation 1 (hvert skridt overvejes) -a Liste over atomer eller anioner artsdannelse -c Liste over kationer artsdannelse – L Bond længde artsdannelse 2 -t Temperatur vibrationer, rheologi -v Diskretisering af bredden af prøveudtagningsvinduet i banen til den gennemsnitlige firkantede forskydningsanalyse Msd 20 -z Diskretisering af starten af prøveudtagningsvinduet i banen til den gennemsnitlige firkantede forskydningsanalyse Msd 20 Tabel 1: De fleste almindelige flag, der bruges i UMD-pakken, og deres mest almindelige betydning. Start med at omdanne outputtet af MD-simuleringen, der udføres i en første-principkode, som VASP8 eller QBox9, til en UMD-fil. Hvis MD-simuleringerne blev udført i VASP, skal du skrive følgende på kommandolinjetypen:VaspParser.py -f -i hvor –f flag definerer navnet på VASP OUTCAR-filen, og -i termiskisering længde.BEMÆRK: Det første trin, defineret af -jeg giver mulighed for at kassere de første trin i simuleringer, som repræsenterer thermalization. I et typisk molekylær-dynamikløb repræsenterer den første del af beregningen termikiseringen, dvs. den tid, det tager systemet for alle atomer at beskrive en gaussisk-lignende fordeling af temperaturen, og for hele systemet at udvise udsving i temperatur, tryk, energi osv. omkring ligevægtsværdier. Denne termiskgørelsesdel af simuleringen bør ikke tages i betragtning ved analyse af væskens statistiske egenskaber. Transformer . umd-filer i . xyz filer til at lette visualisering på forskellige andre pakker, som VMD4 eller Vesta5. På kommandolinjetypen:umd2xyz.py -f -i -s hvor –f definerer navnet på . umd fil, -i definerer thermalization periode, der skal kasseres, og -s hyppigheden af prøveudtagningen af bane gemt i . umd-fil . Standardværdierne er –i 0 -s 1, dvs. Tilbagefør umd-filen til VASP-type POSCAR-filer ved hjælp af umd2poscar.py-scriptet. snapshots af simuleringerne kan vælges med en foruddefineret frekvens. På kommandolinjetypen:umd2poscar.py -f -i -l -s hvor – l repræsenterer det sidste trin, der skal omdannes til POSCAR-fil. Standardværdierne er -i 0 -l 10000000 -s 1. Denne værdi af -l er stor nok til at dække en typisk hele bane. 2. Udfør strukturanalysen Kør gofrs_umd.py scriptet for at beregne PDF-funktionen (Pair Distribution Function) gᴀʙ(r) for alle par af atomtyperne A og B (figur 3). Outputtet er skrevet i én ASCII-fil, tabulatorsepareret, med filtypenavnet gofrs.dat. På kommandolinjetypen:gofrs_umd.py -f -s -d -i BEMÆRK: Standarderne er Sampling_Frequency (hyppigheden for prøveudtagning af bane) = 1 trin; DiscretizationInterval (til plotning af g(r)) = 0,01 Å; InitialStep (antal trin i begyndelsen af den bane, der kasseres) = 0. Den radiale PDF, gᴀʙ(r) er det gennemsnitlige antal atomer af type B i en afstand d_ᴀʙ inden for en sfærisk skal af radius r og tykkelse dr centreret om atomer af type A (figur 3):med ρ atomtætheden, NA og NB antallet af atomer af type A og B, og δ(r−rᴀʙ) deltafunktionen, som er lig med 1, hvis atomer A og B ligger i en afstand mellem r og r +dr. Abscissa af det første maksimum af gᴀʙ(r) giver den højeste sandsynlighedsbindingslængde mellem atomer af type A og B, som er tættest på en gennemsnitlig obligationsafstand, som vi kan bestemme. Det første minimum afgrænser omfanget af det første koordineringsområde. Derfor giver den integrerede del af PDF-filen op til det første minimum det gennemsnitlige koordineringsnummer. Summen af Fourier omdanner af gᴀʙ(r) for alle par af atomare typer A og B giver diffraktion mønster af væsken, som opnås eksperimentelt med et diffraktometer. Men i virkeligheden, som ofte den høje orden koordinering sfærer mangler fra gᴀʙ(r), diffraktion mønster kan ikke opnås i sin helhed. Figur 3: Bestemmelse af parfordelingsfunktion.a) For hvert atom af en art (f.eks. rødt) tælles alle atomer af de koordinerende arter (f.eks. grå og/eller rød) som en funktion af afstanden. (b) Den resulterende afstandsfordelingsgraf for hvert øjebliksbillede, som på dette stadium kun er en samling deltafunktioner, beregnes derefter i gennemsnit over alle atomer og alle snapshots og vægtes af den ideelle gasfordeling for at generere (c) den parfordelingsfunktion, der er kontinuerlig. Det første minimum af g(r) er radius for den første koordinationssfære, der anvendes senere i speciationsanalysen. Klik her for at se en større version af dette tal. Uddrag de gennemsnitlige interatomare bindingsafstande som radier i de første koordinationssfærer. Til dette skal du identificere placeringen af det første maksimum af gᴀʙ(r) funktioner: plot gofrs.dat-filen i et regnearksprogram og søg efter maxima og minima for hvert par atomer. Identificer radius for den første koordinationssfære som det første minimum af PDF-filen , gᴀʙ(r), ved hjælp af regnearkssoftware. Dette er grundlaget for hele den strukturelle analyse af væsken; PDF giver den gennemsnitlige limning status for atomer i væsken. Uddrag afstandene til den første minima, dvs abscissa, og skriv dem i en separat fil, kaldet for eksempel bonds.input. Du kan også køre et af umd-pakkens analyze_gofr scripts for at identificere maxima- og minima-funktionerne i gᴀʙ(r). På kommandolinjetypen:analyze_gofr_semi_automatic.py Klik på placeringen af det maksimale og det mindste af gᴀʙ(r), der vises i den graf, der åbnes af programmet. Scriptet scanner automatisk den aktuelle mappe, identificerer alle gofr.dat filer og udfører analysen for hver enkelt af dem. Klik igen på det maksimale og det mindste i vinduet, hver gang scriptet har brug for en uddannet første gæt. Åbn og se på den automatisk genererede fil kaldet bonds.input , der indeholder de interatomare obligationsafstande. 3. Udfør speciationsanalysen Beregne topologien af limning mellem atomer, ved hjælp af begrebet tilslutningsmuligheder inden for grafteori: atomer er knudepunkter og interatomare obligationer er stierne. Det speciation_umd.py script skal bruge de interatomare bindingsafstande, der er defineret i filen bonds.input .BEMÆRK: Konnektivitetsmatrixen er konstrueret på hvert trin: To atomer, der ligger i en afstand, der er mindre end radiusen af deres tilsvarende første koordinationssfære, anses for at være bundet, dvs. Forskellige atomare netværk er bygget ved at behandle atomer som knudepunkter i en graf, hvis forbindelser er defineret af dette geometriske kriterium. Disse netværk er atomarterne, og deres ensemble definerer atomspektationen i den pågældende væske (figur 4). Figur 4: Identifikation af atomklyngerne.Koordinationen polyhedra er defineret ved hjælp af interatomare afstande. Alle atomer i en afstand, der er mindre end en bestemt radius, anses for at være bundet. Her svarer tærsklen til den første koordinationssfære (de lyse røde cirkler), defineret i figur 1. Polymerisering og dermed kemiske arter er fremstillet af netværk af de bundne atomer. Bemærk den centrale Red1Grey2 klynge, som er isoleret fra de andre atomer, som danner en uendelig polymer. Klik her for at se en større version af dette tal. Kør speciation script for at opnå tilslutning matrix og opnå koordinering polyhedra eller polymerisering. På kommandolinjetypen:speciation_umd.py -f -s -i -l -c -a -m -r hvor -i-flaget giver filen med de interatomare bindingsafstande, som f.eks. blev produceret i det foregående trin. Du kan også køre scriptet med en enkelt længde for alle obligationer, der er defineret af -l-flaget.BEMÆRK: -c flag angiver de centrale atomer, og -en flag ligands. Både centrale atomer og ligands kan være af forskellige typer; i dette tilfælde skal de adskilles af kommaer. -m flaget giver den mindste tid, en art skal leve for at komme i betragtning i analysen. Som standard er denne minimumstid nul, og alle forekomster tælles med i sidste ende. Kør speciation_umd.py scriptet med flaget -r 0, som prøver tilslutningsgrafen på første niveau for at identificere koordinationspolyhedraen. For eksempel kan et centralt atom, der er angivet som en kation , være omgivet af en eller flere anioner (figur 4). Speciation script identificerer hver eneste af koordineringen polyhedra. Det vejede gennemsnit af alle koordinationspolyhedraen giver koordinationsnummeret, identisk med det, der opnås ved integrationen af PDF-filen. På kommandolinjetypen:speciation_umd.py -f -i -c -a -r 0BEMÆRK: Gennemsnitlige koordinationstal i væsker er fraktioneret antal. Denne fraktionerethed kommer fra den gennemsnitlige egenskab ved koordineringen. Definitionen baseret på speciation giver en mere intuitiv og informativ repræsentation af væskens struktur, hvor de relative proportioner af de forskellige arter, dvs. Kør speciation_umd.py script med flaget -r 1, som prøver tilslutningsgrafen på alle dybdeniveauer for at opnå polymeriseringen. Netværket gennem atomgrafen har en vis dybde, da atomer er bundet længere væk til andre bindinger (f.eks. i sekvenser af skiftende kationer og anioner) (figur 4). Åbn de to filer . popul.dat og . stat.dat fortløbende; disse udgør output af speciation script. Hver klynge er skrevet på en linje og angiver dens kemiske formel, det tidspunkt, hvor den dannede, det tidspunkt, hvor den døde, dens levetid, en matrix med listen over de atomer, der danner denne klynge. Plot levetiden for hver atomklynge af alle de kemiske arter, der findes i simuleringen som findes i .popul.dat fil (Figur 5). Plot populationen analyse med overflod af hver art, som findes i . stat.dat fil. Denne analyse, både absolut og relativ, svarer til de faktiske statistikker for koordinationspolyhedraen for sagen -r 0; for polymerisering, med -r 1 dette skal behandles omhyggeligt som nogle normalisering over det relative antal atomer kan være nødvendigt at anvende. Overfloden svarer til den integrerede i løbet af levetiden. Den. stat.dat fil viser også størrelsen af hver klynge, dvs. hvor mange atomer danner det. 4. Beregningsdiffusionskoefficienter Uddrag de gennemsnitlige kvadratiske forskydninger (MSD) af atomer som en funktion af tiden til at opnå selv-diffusivitet. Standardformlen for MSD er:hvor præfaktorerne er renormalisationer. Med MSD-værktøjet er der forskellige måder at analysere de dynamiske aspekter af væskerne på.BEMÆRK: T er den samlede tid for simuleringen og N α er antallet af atomer af typen α. Den første gang t0 er vilkårlig og spænder over den første halvdel af simuleringen. Ninit er antallet af første gange. τ er bredden af det tidsinterval, som MSD beregnes over. dens maksimale værdi er halvdelen af simuleringens tidstid. I typiske MSD-implementeringer starter hvert vindue i slutningen af det foregående. Men en sparsom prøveudtagning kan fremskynde beregningen af MSD, uden at ændre hældningen af MSD. Til dette starter det i-th-vindue på tidspunktet t0(i), men (i+1)-th-vinduet starter på tidspunktet t0(i) + τ + v, hvor værdien af v er brugerdefineret. På samme måde øges vinduets bredde i diskrete trin, der er defineret af brugeren, som sådan: τ(i) = τ(i-1) + z. Værdierne z (“vandret trin”) og v (“lodret trin”) er positive eller nul; Standarden for begge er 20. Beregn MSD ved hjælp af serien af msd_umd scripts. Deres output udskrives i en . msd.dat fil, hvor MSD af hver atomtype, atom eller klynge udskrives på en kolonne som en funktion af tiden. Beregne den gennemsnitlige MSD for hver atomtype. MSD beregnes for hvert atom og derefter gennemsnit for hver atomtype. Outputfilen indeholder én kolonne for hver atomtype. På kommandolinjetypen:msd_umd.py -f -z -v -b Beregne MSD af hvert atom. MSD beregnes for hvert atom og derefter gennemsnit for hver atomtype. Outputfilen indeholder en kolonne for hvert atom i simuleringen og derefter en kolonne for hver atomtype. Denne funktion giver mulighed for at identificere atomer, der diffus i to forskellige miljøer, som væske og gas, eller to væsker. På kommandolinjetypen:msd_all_umd.py -f -z -v -b Beregne den kemiske arts msd. Brug populationen af klynger, der er identificeret med speciationsscriptet, og som udskrives i . popul.dat fil. Msd beregnes for hver enkelt klynge. Outputfilen indeholder én kolonne for hver klynge. For at undgå at overveje store polymerer skal du sætte en grænse for klyngens størrelse; dens standard er 20 atomer. På kommandolinjetypen:msd_cluster_umd.py -f -p -s -b -c BEMÆRK: Standardværdierne er: –b 100 –s 1 –c 20. Plot msd ved hjælp af et regneark-baseret software (Figur 6). I en loglogrepræsentation af MSD i forhold til tid skal du identificere hældningsændringen. Adskil den første del, normalt kort, som repræsenterer det ballistiske regime, dvs. bevarelsen af atomernes hastighed efter kollisioner. Den anden længere del repræsenterer diffusivt regime, dvs. spredning af atomers hastighed efter kollisioner. Beregne diffusionskoefficienterne fra msd-hældningen som:hvor Z er antallet af frihedsgrader (Z = 2 for diffusion i plan, Z = 3 for diffusion i rummet), og t er tidstrinnet. 5. Tidskorrelationsfunktioner Beregning af tidskorrelationsfunktionerne som et mål for systemets inerti ved hjælp af den generelle formel:A kan være en række tidsafhængige variabler, såsom atomare positioner, atomare hastigheder, belastninger, polarisering osv., der hver giver – via Green-Kubo-forbindelserne12,13 – forskellige fysiske egenskaber, nogle gange efter en yderligere transformation. Analysere de atomare hastigheder for at opnå det vibrationelle spektrum af væsken og alternative udtryk for de atomare selvdiffusionskoefficienter. Kør vibr_spectrum_umd.py script til at beregne atomare hastighed-hastighed auto-korrelation (VAC) funktion for hver atomare type og udføre sin hurtige Fourier transformering. På kommandolinjetypen:vibr_spectrum_umd.py -f -t hvor –t er den temperatur, der skal defineres af brugeren. Scriptet udskriver to filer: . vels.scf.dat fil med VAC-funktionen for hver atomtype og . vibr.dat fil med vibrationelle spektrum nedbrudt på hver atomart og den samlede værdi. Åbn og læs vels.scf.dat. Plot VAC-funktionen ud fra vels.scf.dat-filen ved hjælp af regnearkslignende software. Behold den rigtige del af Fourier VAC. Dette er, hvad der giver vibrationelle spektrum, som en funktion af frekvens:hvor m er atommasserne. Plot vibrationelle spektrum fra vibr.dat fil ved hjælp af regneark-lignende software (Figur 7). Identificer den finite værdi ved ω=0, der svarer til væskens diffusive karakter og de forskellige toppe af spektret ved begrænset frekvens. Identificer hver atomtypes deltagelse i vibrationsspektret.BEMÆRK: Nedbrydningen på atomtyper viser, at forskellige atomer har forskellige ω= 0 bidrag, svarende til deres diffusionskoefficienter. Den generelle form af spektret er meget glattere med færre funktioner end for en tilsvarende solid. Ved skallen skal du læse den integrerede over det vibrationelle spektrum, som giver diffusionskoefficienterne for hver atomart.BEMÆRK: Termodynamiske egenskaber kan opnås ved integration fra vibrationsspektret, men resultaterne skal anvendes med forsigtighed på grund af to tilnærmelser: integrationen er gyldig inden for den kvasi-harmoniske tilnærmelse, som ikke nødvendigvis holder ved høje temperaturer; og den gaslignende del af spektret, der svarer til diffusionen, skal kasseres. Integrationen bør så kun ske over den gitterlignende del af spektret. Men denne adskillelse kræver normalt flere yderligere efterbehandlingstrin og beregninger14, som ikke er omfattet af den nuværende UMD-pakke. Kør viscosity_umd.py scriptet for at analysere selvkorrelationen af komponenterne stress tensor at estimere viskositeten af smelten. På kommandolinjetypen:viscosity_umd.py -f -i -s -o -l BEMÆRK: Denne funktion er sonderende, og eventuelle resultater skal tages med forsigtighed. For det første skal viskositetens konvergens grundigt kontrolleres med hensyn til simuleringens længde. Udlede viskositeten af væsken fra selvkorrelationen af stress tensor15 som:hvor V og T er henholdsvis volumen og temperatur, κB er Boltzmann konstant og σ ij ij off-diagonal komponent af stress-tensor, udtrykt i kartesiske koordinater. Brug en mere passende pasform til at opnå et mere robust skøn over viskositeten15,16 og undgå støj fra den stress-tensor autokorrelationsfunktion, der kan opstå som følge af simuleringernes begrænsede størrelse og begrænsede varighed. For den automatiske korrelation funktion af stress tensor, skal du bruge følgende funktionelle form15,16, der giver gode resultater:hvor A, B, τ1, τ2 og ω er fit parametre. Efter integration bliver udtrykket for viskositeten: 6. Termodynamiske parametre, der stammer fra simuleringerne. Kør averages.py for at udtrække gennemsnitsværdierne og spredningen (som standardafvigelse) for tryk, temperatur, tæthed og intern energi fra umd-filerne . På kommandolinjetypen:averages.py -f -s med –s 0 som standard. Beregn den statistiske fejl i gennemsnittet ved hjælp af blokeringsmetoder.BEMÆRK: Der er forskellige varianter af denne metode. Efter arbejdet i Allen og Tildesley2, er det almindeligt at gennemsnittet over sekvenser af tidsblokke, af stadig længere længde, og anslå standardafvigelsen med hensyn til det aritmetiske gennemsnit17. Konvergens kan opnås inden for grænsen af mange- og længe nok blokstørrelser, når prøveudtagningen ikke er korreleret. Selv om den faktiske tærskelværdi for konvergensen normalt skal vælges manuelt. Brug halveringsmetoden18: Start med den første dataprøve, ved hvert trin κ, halvere antallet af prøverne ved i gennemsnit over hver to tilsvarende på hinanden følgende prøver fra det foregående trin κ−1: Kør fullaverages.py scriptet for at udføre den komplette statistiske analyse, herunder middelværdiens fejl. På kommandolinjetypen:fullaverages.py BEMÆRK: Scriptet automatiseres til det punkt, hvor der søges efter alle .umd.dat filer i den aktuelle mappe og udfører analysen for dem alle. Standardindstillingerne er –s 0 –u 0. For -u 0 output er minimal, og for -u 1 output er i fuld, med flere alternative enheder udskrevet. Dette script kræver grafisk understøttelse, da det skaber et grafisk billede til kontrol af konvergensen til estimering af fejlen på middelværdien.