Questo protocollo descrive un metodo di throughput medio-alto per lo screening di librerie di frammenti utilizzando alcuni strumenti di robotica e misurazione comuni. Schermate come quelle descritte in questo protocollo possono essere eseguite di routine dalla NKI Protein Facility di Amsterdam, ad esempio come servizio iNEXT-Discovery, spesso anche gratuitamente per gli utenti dopo l’applicazione della proposta e la revisione tra pari. In questi casi, la libreria DSi-Poised può essere fornita dalla struttura, ma l’uso di altre librerie può anche essere discusso nel contesto di ogni diversa applicazione utente e contratto di servizio. La scelta degli strumenti in questo protocollo rappresenta soluzioni pratiche per molti laboratori, ma non deve essere considerata come un gold standard. Per misurare la stabilità termica della proteina bersaglio per lo screening dei frammenti sono raccomandati metodi privi di etichette per misurare la stabilità termica della proteina bersaglio, piuttosto che metodi che utilizzano etichette sensibili all’ambiente per rilevare lo svolgimento in un termociclatore a reazione a catena della polimerasi a trascrizione inversa.
I metodi senza etichetta, come quello qui presentato con lo strumento Prometheus, hanno alcuni vantaggi: usano basse quantità di proteine, spesso un paio di ordini di grandezza in meno; possono essere utilizzati per misurare simultaneamente lo scattering del campione e quindi l’aggregazione; e le etichette utilizzate per rilevare lo svolgimento in altri approcci possono interagire in modo diverso con ciascun frammento, con conseguenti artefatti di misurazione. Questo protocollo è stato descritto nel contesto del robot Mosquito, che consente il pipettaggio di un volume molto piccolo di campione (0,3 μL) che non può essere fatto manualmente. Il Mosquito è un robot popolare, presente in molti laboratori che lavorano su progetti di biologia strutturale e scoperta di farmaci; tuttavia, il protocollo può chiaramente utilizzare approcci alternativi per il pipettaggio a basso volume.
Le librerie di frammenti contengono composti disciolti in DMSO. Una delle sfide iniziali è trovare la concentrazione ottimale di DMSO alla quale la proteina rimane stabile e i composti rimangono solubili. Ciò comporta l’esecuzione delle misurazioni a varie concentrazioni di DMSO per determinare le condizioni ottimali per lo screening. La diluizione da proteina a frammento qui utilizzata si traduce in concentrazioni di DMSO dello 0,2%; la maggior parte delle proteine sono abbastanza stabili in queste condizioni. La quantità di proteine necessaria per effettuare lo screening per la libreria di 768 composti è di ~ 2-3 mg in totale, poiché le misurazioni vengono in genere eseguite a basse concentrazioni proteiche (0,2 mg mL-1). Lavorare con concentrazioni proteiche relativamente basse non solo riduce i costi di produzione delle proteine, ma riduce anche le possibilità di precipitazione delle proteine. La bassa concentrazione proteica non influisce sul rilevamento del legame dei frammenti, poiché la concentrazione dei frammenti nell’esperimento è di ~ 2 mM, consentendo di identificare anche i leganti deboli.
Poiché il rilevamento della transizione di fusione in questi esperimenti si basa sull’intensità della fluorescenza, un aspetto critico è determinare il potere di eccitazione del laser a cui effettuare le misurazioni. L’interazione dei composti con la proteina può (i) non avere alcun effetto sulla sua fluorescenza intrinseca, (ii) provocare la tempra o (iii) aumentare la sua fluorescenza intrinseca. Inoltre, lavorare con basse concentrazioni proteiche significa che il conteggio di fluorescenza per la proteina nativa sarebbe basso. Il potere di eccitazione deve quindi essere regolato in modo tale da poter misurare la maggior parte dei campioni. Il profilo di scattering di ogni esecuzione fornisce informazioni importanti sugli effetti di aggregazione che potrebbero essere attivati dall’aggiunta di qualsiasi frammento. Inoltre, l’effetto della temperatura sulla solubilità del composto può essere visto anche sul profilo di scattering.
Inaspettatamente, per molti composti è stato osservato che lo scattering in realtà diminuiva con l’aumentare della temperatura (Figura 6). È quindi importante esaminare sia la curva di transizione di fusione che il profilo di scattering di accompagnamento per decidere l’affidabilità di ciascun esperimento, specialmente per quei frammenti che sono considerati candidati per misurazioni più impegnative mediante cristallografia a raggi X o spettroscopia NMR, o addirittura considerati come hit per la chimica di follow-up. Una limitazione specifica del metodo ai fini dello screening dei frammenti è che molti frammenti nella libreria DSi-Poised hanno una fluorescenza intrinseca significativa, a volte anche oltre il limite di saturazione del rivelatore, e quindi questi non possono essere adeguatamente sottoposti a screening per il legame target anche a basso potere di eccitazione. Un altro punto da notare per questo metodo è che può essere utilizzato solo con proteine contenenti residui di triptofano.
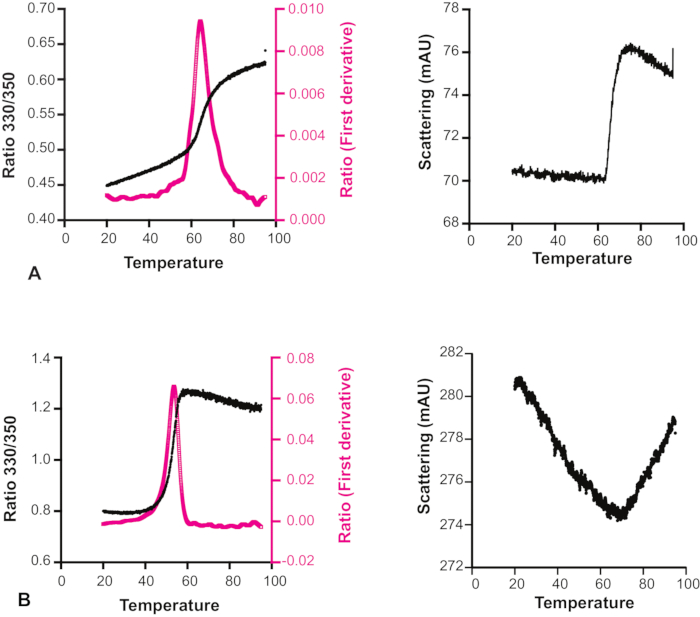
Figura 6: Effetto della temperatura sulla solubilità del composto. Curva di transizione di fusione e profilo di scattering di Hec1 con due composti diversi. (A) Il profilo di dispersione mostra che per questo campione la solubilità non è influenzata dalla temperatura. (B) Il profilo di dispersione mostra che la solubilità di questo campione aumenta con l’aumentare della temperatura. La curva di transizione di fusione in questo caso non è quindi affidabile. Fare clic qui per visualizzare una versione più grande di questa figura.
Una domanda aperta è cosa dovrebbe essere considerato come un cambiamento significativo in Tm, non da una prospettiva matematica, ma dal punto di vista pratico: quale cambiamento in Tm è importante considerare come indicativo del legame di un ligando a una proteina? In questi esempi, si vedono spostamenti inferiori a 1 °C per il 74% dei frammenti per legare Hec1, il 66% per Mps1 e il 53% per Nsp5. Considerare 1 °C come “cambiamento significativo” difficilmente fornirebbe risultati degni di essere perseguiti dalla chimica di follow-up. Nei grafici di panoramica (Figura 5), sono stati considerati contenitori di 1, 2, 5 o più di 5 gradi di spostamento Tm, sia positivi che negativi. Ciò richiede modifiche in base a ciascun caso specifico per fornire una buona panoramica e consentire un processo decisionale informato, determinando il passo successivo. In particolare, per alcune proteine, sono state osservate sia la stabilizzazione che la destabilizzazione del bersaglio a seconda dei frammenti considerati. Entrambi gli eventi sono interessanti, in quanto entrambi possono essere il risultato del legame dei frammenti, ed entrambi possono portare a una buona molecola di follow-up per manipolare il comportamento delle proteine.
Rimane un’ultima domanda, vale a dire, “cosa definisce un colpo utile?”. In realtà, la risposta dipende dalla situazione specifica. Ad esempio, per Hec1, tutti i frammenti che stabilizzano la proteina di oltre 2 gradi o la destabilizzano di più di 5 sono stati comunicati ai nostri collaboratori di chimica, che hanno progettato nuove molecole basate su questi colpi. Per Nsp5, tuttavia, i colpi più destabilizzanti sono stati comunicati ai nostri collaboratori NMR per confermare i colpi derivati dal nanoDSF con esperimenti NMR. In altre parole, i risultati dello screening ottenuti da questo protocollo dovrebbero essere analizzati con cautela e in modo dipendente dal contesto, prendendo decisioni informate basate sulla domanda specifica e sulla metodologia circostante. In ogni caso, il metodo qui descritto è un approccio complementare alle metodologie esistenti come lo screening a raggi X e nmR, che può mirare a confermare, dare priorità o dare nuove idee per le campagne di chimica.
Tabella supplementare S1: Elenco dei tamponi utilizzati per lo screening delle proteine. Abbreviazioni: HEPES = acido 4-(2-idrossietil)-1-piperazineetansolfonico; DTT = ditiotritolo; MOBS = acido 4-(N-morfolino)butanisolfonico. Fare clic qui per scaricare questa tabella.
Tabella supplementare S2: Proprietà per la piastra MRC a 2 pozzi per l’uso con robot nanodispenser. Fare clic qui per scaricare questa tabella.
Tabella supplementare S3: Proprietà per piastra con fondo a V a 96 pozzi per l’uso con robot nanodispenser. Fare clic qui per scaricare questa tabella.
Tabella supplementare S4: Il tampone, la concentrazione proteica ela Tm delle proteine discusse nei risultati rappresentativi. Abbreviazioni: DTT = ditiotritolo; Hec1 = Altamente espresso nella proteina Cancer 1; Mps1 = chinasi monopolare del fuso 1; Nsp5 = proteasi simile a SARS-CoV-2 3C. Fare clic qui per scaricare questa tabella.
File supplementare 1: Parametri di panoramica–datidi esempio. Fare clic qui per scaricare questo file.
File supplementare 2: valori Tm e ΔTm per 406 frammenti-dati di esempio. Fare clic qui per scaricare questo file.
