Preparazione del campione all'analisi bioinformatica della metilazione del DNA: strategia di associazione per l'obesità e studi sui tratti correlati
Summary
Il presente studio descrive il flusso di lavoro per gestire i dati di metilazione del DNA ottenuti dalle tecnologie microarray. Il protocollo illustra i passaggi dalla preparazione del campione all’analisi dei dati. Tutte le procedure sono descritte in dettaglio e il video mostra i passaggi significativi.
Abstract
L’obesità è direttamente collegata allo stile di vita ed è stata associata a cambiamenti di metilazione del DNA che possono causare alterazioni nell’adipogenesi e nei processi di stoccaggio dei lipidi che contribuiscono allo sviluppo della malattia. Dimostriamo un protocollo completo dalla selezione all’analisi dei dati epigenetici di pazienti con e senza obesità. Tutte le fasi del protocollo sono state testate e convalidate in uno studio pilota. 32 donne hanno partecipato allo studio, in cui 15 individui sono stati classificati con obesità in base all’indice di massa corporea (BMI) (45,1 ± 5,4 kg / m2); e 17 individui sono stati classificati senza obesità in base al BMI (22,6 ± 1,8 kg / m2). Nel gruppo con obesità, 564 siti CpG correlati alla massa grassa sono stati identificati mediante analisi di regressione lineare. I siti CpG erano nelle regioni promotrici. L’analisi differenziale ha rilevato 470 CpG ipometilati e 94 siti ipermetilati in individui con obesità. Le vie arricchite più ipometilate erano nel RUNX, nella segnalazione WNT e nella risposta all’ipossia. Le vie ipermetilate erano correlate alla secrezione di insulina, alla segnalazione del glucagone e al Ca2+. Concludiamo che il protocollo ha identificato efficacemente i modelli di metilazione del DNA e la metilazione del DNA correlata ai tratti. Questi modelli potrebbero essere associati a un’alterata espressione genica, influenzando l’adipogenesi e l’accumulo di lipidi. I nostri risultati hanno confermato che uno stile di vita obesogenico potrebbe promuovere cambiamenti epigenetici nel DNA umano.
Introduction
Le tecnologie omiche su larga scala sono state sempre più utilizzate negli studi sulle malattie croniche. Una caratteristica interessante di questi metodi è la disponibilità di una grande quantità di dati generati per la comunità scientifica. Pertanto, è sorta la richiesta di standardizzare i protocolli per consentire il confronto tecnico tra gli studi. Il presente studio suggerisce la standardizzazione di un protocollo per ottenere e analizzare i dati di metilazione del DNA, utilizzando uno studio pilota come esempio applicato.
Il dispendio energetico negativo predomina nei moderni stili di vita umani, portando ad un eccessivo accumulo di tessuto adiposo e, di conseguenza, allo sviluppo dell’obesità¹. Molti fattori hanno aumentato i tassi di obesità, come il sedentarismo, le diete ipercaloriche e le routine stressanti. L’Organizzazione Mondiale della Sanità (OMS) ha stimato che 1,9 miliardi di adulti erano obesi nel 2016, il che significa che oltre il 20% della popolazione mondiale ha oltre 30 kg / m2 BMI2. L’aggiornamento più recente del 2018 ha rivelato che la prevalenza dell’obesità negli Stati Uniti d’America (USA) era superiore al 42%3.
L’epigenetica è l’adattamento strutturale delle regioni cromosomiche per registrare, segnalare o perpetuare stati di attività alterati4. La metilazione del DNA è un’alterazione chimica reversibile nei siti dei dinucleotidi citosina-guanosina (siti CpG), formando 5-metilcitosina-pG (5mCpG). Può modulare l’espressione genica regolando l’accesso del meccanismo di trascrizione al DNA5,6,7,8. In questo contesto, è essenziale capire quali siti CpG sono associati a tratti correlati all’obesità9. Molti fattori possono supportare o prevenire la metilazione del DNA sito-specifica. Gli enzimi necessari per questo processo, come le DNA metiltransferasi10 (DNTM) e le traslocazioni di dieci-undici (TET), possono promuovere la metilazione o la demetilazione del DNA in caso di esposizioni ambientali11.
Considerando il crescente interesse per gli studi di metilazione del DNA negli ultimi anni, la scelta della strategia di analisi più appropriata per rispondere con precisione a ciascuna domanda è stata una preoccupazione essenziale dei ricercatori12,13,14. L’array di metilazione del DNA 450K è il metodo più popolare, utilizzato in oltre 360 pubblicazioni14 per determinare il profilo di metilazione del DNA. Può determinare la metilazione di un massimo di 485.000 CpG situati nel 99% dei geni noti15. Tuttavia, questo array è stato interrotto e sostituito con EPIC, coprendo 850.000 siti CpG. Il presente protocollo può essere applicato sia per 450K che per EPIC16,17,18.
Il protocollo è presentato passo dopo passo nella Figura 1 e comprende le seguenti fasi: selezione della popolazione, campionamento, preparazione dell’esperimento, pipeline di metilazione del DNA e analisi bioinformatica. Uno studio pilota eseguito nel nostro laboratorio è qui dimostrato per illustrare i passaggi del protocollo proposto.
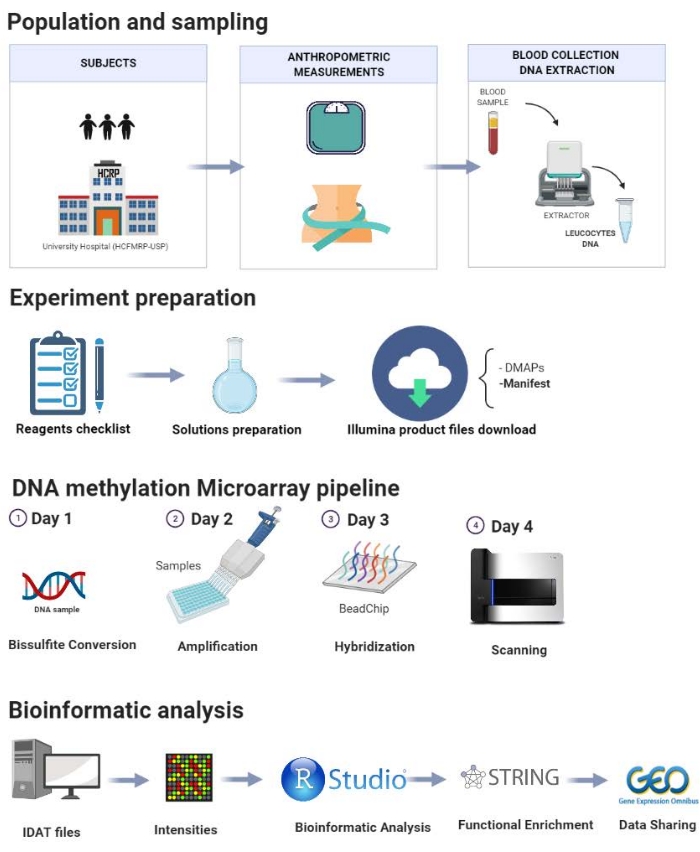
Figura 1: Schema del protocollo presentato. Fare clic qui per visualizzare una versione più grande di questa figura.
Protocol
Representative Results
Discussion
Gli array di metilazione del DNA sono i metodi più utilizzati per accedere alla metilazione del DNA grazie al loro rapporto costi-benefici14. Il presente studio ha descritto un protocollo dettagliato che utilizza una piattaforma di microarray disponibile in commercio per valutare la metilazione del DNA in uno studio pilota eseguito in una coorte brasiliana. I risultati ottenuti dallo studio pilota hanno confermato l’efficacia del protocollo. La figura 3 mostra la comparabilità del campione e la conversione completa del bisolfito32.
Come fase di controllo qualità, l’algoritmo ChAMP ha raccomandato l’esclusione dei siti CpG durante il processo di filtraggio. L’obiettivo di escludere le sonde è migliorare l’analisi dei dati ed eliminare i pregiudizi. I CpG di bassa qualità (valori p inferiori a 0,05) sono stati rimossi per eliminare il rumore sperimentale nel set di dati. Gli obiettivi sono rimasti superati nell’analisi del grafico di densità. Zhou33 ha descritto l’importanza di filtrare i CpG vicino agli SNP per evitare discrepanze, interpretazioni errate della metilazione delle citosine polimorfiche e causare il colore dell’interruttore del design della sonda di tipo I34. Inoltre, poiché i cromosomi XY sono influenzati in modo differenziale dall’imprinting, Heiss e Just35 hanno rafforzato l’importanza di filtrare tali sonde perché, nelle femmine, i problemi con l’ibridazione possono essere fattori confondenti35.
La data di scadenza dei DMAP, la data di apertura della formammide, la qualità analitica dell’etanolo assoluto e la conta totale dei leucociti sono considerate fasi critiche nel protocollo.
Inoltre, secondo le nostre osservazioni, la stima del tipo di cellula è essenziale per eseguire l’analisi bioinformatica. Il metodo Houseman esegue la stima del tipo di cellula come descritto nello studio di Tian30. Questo metodo si basa su 473 siti CpG specifici in grado di prevedere le percentuali dei tipi di cellule più importanti, come granulociti, monociti, cellule B e cellule T36. Abbiamo usato la funzione consigliata “myRefbase” dal pacchetto ChAMP. Dopo la stima, l’algoritmo ChAMP regola i valori beta ed elimina questa distorsione dal set di dati. Questo passaggio è cruciale negli studi incentrati sull’obesità perché questa popolazione ha una notevole differenza nei globuli bianchi a causa del loro stato infiammatorio cronico.
Abbiamo solo modificato la mappa del cappuccio originale per il sigillo PCR comune per quanto riguarda la modifica del metodo e la risoluzione dei problemi. Dopo ogni processo di centrifugazione, il sigillo è stato cambiato con uno nuovo. Non abbiamo potuto utilizzare la termosaldatura standard e l’abbiamo adattata usando un foglio di alluminio attorno alla piastra.
Sebbene i saggi commerciali siano stati considerati un gold standard per gli studi epigenetici, una limitazione del protocollo potrebbe essere la specificità dei reagenti e delle apparecchiature di un marchio unico37,38,39,40. Un’altra limitazione è la mancanza di indicatori che consentano di identificare il corretto svolgimento dell’esperimento41.
La standardizzazione del presente protocollo rappresenta un’ottima guida per la ricerca epigenetica, riducendo gli errori umani durante il processo e consentendo un’analisi dei dati di successo e la comparabilità tra diversi studi.
Secondo i nostri risultati, gli esperimenti di metilazione del DNA sono adatti per studi che confrontano individui con e senza obesità43. Inoltre, l’analisi bioinformatica proposta ha fornito dati di alta qualità e potrebbe essere presa in considerazione in studi su larga scala.
Utilizzando l’analisi SVD, abbiamo identificato che i tratti correlati all’obesità (BMI, WC e FM) hanno influenzato la variabilità nei dati di metilazione del DNA. Come risultato significativo, la stima del tipo di cellula indica che sia le cellule natural killer (NK) che le cellule B erano più alte nelle donne con obesità rispetto alle donne senza obesità (Figura 5). I conteggi più elevati di queste cellule potrebbero essere spiegati dallo stato infiammatorio di basso grado di questi individui44. Abbiamo osservato che i pazienti con obesità hanno CpG ipo- e ipermetilati nelle regioni promotrici dei geni associati alla massa grassa. La maggior parte dei siti erano ipometilati, il che potrebbe essere correlato al naturale aumento dei livelli di specie reattive dell’ossigeno (ROS) in questi individui. Questa condizione di stress ossidativo può promuovere la perturbance della guanina nel sito di dinucleotide, formando 8-idrossi-2′-deossiguanosina (8-OHdG), con conseguente sito dinucleotide 5mCp-8-OHdG e causando il reclutamento degli enzimi TET. Tutti questi eventi potrebbero essere responsabili della promozione dell’ipometilazione e dell’ipermetilazione del DNA da parte di diversi meccanismi d’azione45.
Inoltre, il tasso di adipogenesi sembra aumentare negli individui con obesità, con circa il 10% di cellule nuove a cellule vecchie46,47. I contributi epigenetici, enfatizzando l’ambiente obesogenico, possono alterare i tassi di proliferazione e differenziazione delle cellule, favorendo lo sviluppo della massa grassa48. I cambiamenti epigenetici possono anche influenzare i programmi adipogenici, facilitando o limitando il loro sviluppo. I fattori di trascrizione primaria (PPARγ o C / EBPα) o l’assemblaggio di complessi multiproteici, posizionati in regioni promotori a valle operate includendo o escludendo enzimi modificanti epigenetici, regolano l’espressione genica attraverso iper- o ipometilazione45. La via PPARγ è stata precedentemente descritta per alterare la via WNT, che aveva geni arricchiti in questo studio. Sebbene non sia ancora noto come si verifichi la segnalazione WNT durante l’adipogenesi, studi recenti hanno riportato che potrebbe avere ruoli essenziali nel metabolismo degli adipociti, in particolare in condizioni obesogene49.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Vorremmo ringraziare Yuan Tian, Ph.D. (tian.yuan@ucl.ac.uk) per essere disponibile a rispondere a tutti i dubbi sul pacchetto ChAMP. Ringraziamo anche Guilherme Telles, Msc. per il suo contributo sia alle questioni tecniche che scientifiche di questo articolo; ha fatto importanti considerazioni riguardanti l’epigenetica e le tecniche di cattura e formattazione video (guilherme.telles@usp.br). Finanziamento dei materiali di consumo: Fondazione di ricerca di San Paolo (FAPESP) (# 2018/ 24069-3) e Consiglio nazionale per lo sviluppo scientifico e tecnologico (CNPq: #408292/2018-0). Finanziamento personale: (FAPESP: #2014/16740-6) e programma di eccellenza accademica dal coordinamento per lo sviluppo del personale dell’istruzione superiore (CAPES: 88882.180020 / 2018-01). I dati saranno resi pubblici e liberamente disponibili senza restrizioni. Indirizza la corrispondenza a NYN (e-mail: nataliayumi@usp.br) o CBN (e-mail: carla@fmrp.usp.br).
Materials
| Absolute ethanol | J.T. Baker | B5924-03 | |
| Agarose gel | Kasvi | K9-9100 | |
| Electric bioimpedance | Quantum BIA 450 Q – RJL System | ||
| Ethylenediaminetetraacetic acid (EDTA) | Corning | 46-000-CI | |
| EZ DNA Methylation-Gold kit | ZymoResearch, Irvine, CA, USA | D5001 | |
| Formamide | Sigma | F9037 | |
| FMS—Fragmentation solution | Illumina | 11203428 | Supplied Reagents |
| HumanMethylation450 BeadChip | Illumina | ||
| Maxwell Instrument | Promega, Brazil | AS4500 | |
| MA1—Multi-Sample Amplification 1 Mix | Illumina | 11202880 | Supplied Reagents |
| MicroAmp Optical Adhesive Film | Thermo Fisher Scientific | 201703982 | |
| MSM—Multi-Sample Amplification Master Mix | Illumina | 11203410 | Supplied Reagents |
| NaOH | F. MAIA | 114700 | |
| PB1—Reagent used to prepare BeadChips for hybridization | Illumina | 11291245 | Supplied Reagents |
| PB2—Humidifying buffer used during hybridization | Illumina | 11191130 | Supplied Reagents |
| 2-propanol | Emsure | 10,96,34,01,000 | |
| RA1—Resuspension, hybridization, and wash solution | Illumina | 11292441 | Supplied Reagents |
| RPM—Random Primer Mix | Illumina | 15010230 | Supplied Reagents |
| STM—Superior Two-Color Master Mix | Illumina | 11288046 | Supplied Reagents |
| TEM—Two-Color Extension Master Mix | Illumina | 11208309 | Supplied Reagents |
| Ultrapure EDTA | Invitrogen | 155576-028 | |
| 96-Well Reaction Plate with Barcode (0.1mL) | ByoSystems | 4346906 | |
| 96-Well Reaction Plate with Barcode (0.8mL) | Thermo Fisher Scientific | AB-0859 | |
| XC1—XStain BeadChip solution 1 | Illumina | 11208288 | Supplied Reagents |
| XC2—XStain BeadChip solution 2 | Illumina | 11208296 | Supplied Reagents |
| XC3—XStain BeadChip solution 3 | Illumina | 11208392 | Supplied Reagents |
| XC4—XStain BeadChip solution 4 | Illumina | 11208430 | Supplied Reagents |
References
- Manna, P., Jain, S. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metabolic Syndrome and Related Disorders. 13 (10), 423-444 (2015).
- . Obesity Available from: https://www.who.int/health-topics/obesity#tab=tab_1 (2017)
- Adult Obesity Facts. Center for Disease Control and Prevention Available from: https://www.cdc.gov/obesity/data/adult.html (2021)
- Bird, A. Perceptions of epigenetics. Nature. 396, (2007).
- Kouzarides, T. Chromatin modifications, and their function. Cell. 128 (4), 693-705 (2007).
- Berger, F. The strictest usage of the term epigenetic. Seminars in Cell & Developmental Biology. 19 (6), 525-526 (2008).
- Henikoff, S., Greally, J. M. Epigenetics, cellular memory, and gene regulation. Current Biology. 26 (14), 644-648 (2016).
- Laker, R. C., et al. Transcriptomic and epigenetic responses to short-term nutrient-exercise stress in humans. Scientific Reports. 7 (1), 1-12 (2017).
- Wahl, S., et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 541, 81-86 (2017).
- Jin, B., Robertson, K. D. DNA methyltransferases, DNA damage repair, and cancer. Epigenetic Alterations in Oncogenesis. 754, 3-29 (2013).
- Jang, H. S., Shin, W. J., Lee, J. E., Do, J. T. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes. 8 (6), 148 (2017).
- Shen, L., Waterland, R. A. Methods of DNA methylation analysis. Current Opinion in Clinical Nutrition & Metabolic Care. 10 (5), 576-581 (2007).
- Olkhov-Mitsel, E., Bapat, B. Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers. Cancer Medicine. 1, 237-260 (2012).
- Kurdyukov, S., Bullock, M. DNA methylation analysis: choosing the right method. Biology. 5 (1), 3 (2016).
- Yong, W. -. S., Hsu, F. -. M., Chen, P. -. Y. Profiling genome-wide DNA methylation. Epigenetics & Chromatin. 9 (1), 1-16 (2016).
- Dugué, P. A., et al. Alcohol consumption is associated with widespread changes in blood DNA methylation: Analysis of cross-sectional and longitudinal data. Addiction Biology. 1, 12855 (2021).
- Colicino, E., et al. Blood DNA methylation sites predict death risk in a longitudinal study of 12, 300 individuals. Aging. 14, 14092-14124 (2020).
- Karlsson, L., Barbaro, M., Ewing, E., Gomez-Cabrero, D., Lajic, S. Genome-wide investigation of DNA methylation in congenital adrenal hyperplasia. The Journal of Steroid Biochemistry and Molecular Biology. 201, 105699 (2020).
- Ribeiro, R. R., Guerra-Junior, G., de Azevedo Barros-Filho, A. Bone mass in schoolchildren in Brazil: the effect of racial miscegenation, pubertal stage, and socioeconomic differences. Journal of Bone and Mineral Metabolism. 27 (4), 494-501 (2009).
- Filozof, C., et al. Obesity prevalence and trends in Latin-American countries. Obesity Reviews. 2 (2), 99-106 (2001).
- Chadid, S., Kreger, B. E., Singer, M. R., Bradlee, M. L., Moore, L. L. Anthropometric measures of body fat and obesity-related cancer risk: sex-specific differences in Framingham Offspring Study adults. International Journal of Obesity. 44 (3), 601-608 (2020).
- Nicoletti, C. F., et al. DNA methylation pattern changes following a short-term hypocaloric diet in women with obesity. European Journal of Clinical Nutrition. 74 (9), 1345-1353 (2020).
- Assessing Your Weight. Centers for Disease Control and Prevention Available from: https://www.cdc.gov/healthyweight/assessing/index.html (2020)
- Giavarina, D., Lippi, G. Blood venous sample collection: Recommendations overview and a checklist to improve quality. Clinical Biochemistry. 50 (10-11), 568-573 (2017).
- Duijs, F. E., Sijen, T. A rapid and efficient method for DNA extraction from bone powder. Forensic Science International: Reports. 2, 100099 (2020).
- . Infinium HD Methylation Assay, Manual Protocol Available from: https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/infinium_assays/infinium_hd_methylation/infinium-hd-methylation-guide-15019519-01.pdf (2015)
- Serrano, J., Snuderl, M. Whole genome DNA methylation analysis of human glioblastoma using Illumina BeadArrays. Glioblastoma. , 31-51 (2018).
- Leti, F., Llaci, L., Malenica, I., DiStefano, J. K. Methods for CpG methylation array profiling via bisulfite conversion. Disease Gene Identification. 1706, 233-254 (2018).
- Noble, A. J., et al. A validation of Illumina EPIC array system with bisulfite-based amplicon sequencing. PeerJ. 9, 10762 (2021).
- Tian, Y., et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 33, 3982-3984 (2017).
- Turinsky, A. L., et al. EpigenCentral: Portal for DNA methylation data analysis and classification in rare diseases. Human Mutation. 41 (10), 1722-1733 (2020).
- . The Chip Analysis Methylation Pipeline Available from: https://www.bioconductor.org/packages/release/bioc/vignettes/ChAMP/inst/doc/ChAMP.html (2020)
- Zhou, W., Laird, P. W., Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Research. 45 (4), 22 (2017).
- Wanding, Z., Triche, T. J., Laird, P. W., Hui, S. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Research. 46 (20), 123 (2018).
- Heiss, J. A., Just, A. C. Improved filtering of DNA methylation microarray data by detection p values and its impact on downstream analyses. Clinical Epigenetics. 11, 15 (2019).
- Houseman, E. A., et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 13, 86 (2012).
- Valavanis, I., Sifakis, E. G., Georgiadis, P., Kyrtopoulos, S., Chatziioannou, A. A. A composite framework for the statistical analysis of epidemiological DNA methylation data with the Infinium human Methylation 450K BeadChip. IEEE Journal of Biomedical and Health Informatics. 18 (3), 817-823 (2014).
- Sun, N., Zhang, J., Zhang, C., Shi, Y., Zhao, B., Jiao, A., Chen, B. Using Illumina Infinium HumanMethylation 450K BeadChip to explore genomewide DNA methylation profiles in a human hepatocellular carcinoma cell line. Molecular Medicine Reports. 18 (5), 4446-4456 (2018).
- Moran, S., Arribas, C., Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 8 (3), 389-399 (2016).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Lehne, B., et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biology. 16 (1), 1-12 (2015).
- Wang, J., Zhang, H., Rezwan, F. I., Relton, C., Arshad, S. H., Holloway, J. W. Pre-adolescence DNA methylation is associated with BMI status change from pre- to post-adolescence. Clinical Epigenetics. 25, 64 (2021).
- Maugeri, A. The effects of dietary interventions on DNA methylation: implications for obesity management. International Journal of Molecular Sciences. 21, 8670 (2020).
- DeFuria, J., et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proceedings of the National Academy of Sciences. 110 (13), 5133-5138 (2013).
- Kietzmann, T., Petry, A., Shvetsova, A., Gerhold, J. M., Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. British Journal of Pharmacology. 174, 1533-1554 (2017).
- Chase, K., Sharma, R. P. Epigenetic developmental programs and adipogenesis: implications for psychotropic induced obesity. Epigenetics. 8 (11), 1133-1140 (2013).
- Spalding, K. L., et al. Dynamics of fat cell turnover in humans. Nature. 453, 783-787 (2008).
- Ross, S. E., et al. Inhibition of adipogenesis by Wnt signaling. Science. 289, 950-953 (2000).
- Bagchi, D. P., et al. Wnt/β-catenin signaling regulates adipose tissue lipogenesis and adipocyte-specific loss is rigorously defended by neighboring stromal-vascular cells. Molecular Metabolism. 42, 101078 (2020).
