All animal experiments conformed to national and institutional guidelines, including the Council Directive 2010/63/EU of the European Parliament, and had full Home Office ethical approval (University of Heidelberg Animal Welfare Office and Regierungspraesidium Karlsruhe, licenses T14/21 and T13/21). Primary hippocampal and cortical neurons were prepared from newborn mouse or rat pups according to standard procedures and were maintained for 12-14 days as previously described13.
1. Preparation of solutions
- Stock solutions for imaging buffer
- Prepare each stock solution according to Table 1 and keep them at 4 °C. For long-term storage (>3 months), keep aliquots at -20 °C.
| Component | MW | Concentration (M) | Amount (g) | Volume (mL) |
| NaCl | 58.44 | 5 | 14.61 | 50 |
| KCl | 74.55 | 3 | 1.12 | 5 |
| MgCl2·6H2O | 203.3 | 1.9 | 2 | 5 |
| CaCl2·2H2O | 147.01 | 1 | 1.47 | 10 |
| Glycine | 75.07 | 0.1 | 0.375 | 50 |
| Sucrose | 342.3 | 1.5 | 25.67 | 50 |
| Sodium pyruvate | 110.04 | 0.1 | 0.55 | 50 |
| HEPES | 238.3 | 1 | 11.9 | 50 |
| Glucose | 180.15 | 2.5 | 45 | 100 |
Table 1: Stock solutions for imaging buffer.
- Stock solutions of drugs and dyes
- Dissolve diamide (DA; used for calibration of maximal 405:488 ratio) in water to obtain a 0.5 M stock solution (e.g., 1 g in 11.615 mL of water). Aliquot and store at -20 °C.
- Dissolve dithiothreitol (DTT; used for calibration of minimal 405:488 ratio) in water to obtain a 1 M stock solution (e.g., 5 g in 32.425 mL of water). Aliquot and store at -20 °C for a maximum of 3 months.
- Dissolve N-methyl-D-aspartate (NMDA; used to induce excitotoxicity and mitochondrial oxidation) in water to obtain a 10 mM stock solution (e.g., 25 mg in 16.991 mL of water). Store the aliquots at -20 °C. For long-term storage (>6 months), keep the aliquots at -80 °C.
- Tetramethylrhodamine ethyl ester perchlorate (TMRE; a small-molecule indicator of the mitochondrial membrane potential)
- Dissolve TMRE powder in methanol to obtain a 20 mM stock (e.g., 25 mg in 2.427 mL of methanol).
- Dilute the 20 mM stock 1:1,000 in methanol to obtain a 20 µM stock.
- Aliquot the 20 mM and 20 µM stock solutions, seal with parafilm, and store protected from light at -20 °C.
NOTE: Both stock solutions are stable for several years. Use the 1,000x stock solution (20 µM) for experiments.
- Imaging buffer
- Prepare 100 mL of imaging buffer by adding all components from Table 2 to 80 mL of sterile water in a measuring cylinder. Bring the volume up to 100 mL with sterile water. Mix by carefully shaking the measuring cylinder until the solution appears homogeneous.
NOTE: It is recommended to use an osmometer to check the osmolarity of the buffer. It should be as close as possible to the growth medium of the cells. Here, this is 315 mOsmol/L. Increase or decrease the sucrose concentration as needed to match the osmolarity of the imaging buffer and growth medium. - Adjust the pH to 7.4. Make aliquots and keep them at 4 °C for up to two weeks. For long-term storage, keep the aliquots at -20 °C. Let the imaging buffer reach room temperature before use.
- Prepare 100 mL of imaging buffer by adding all components from Table 2 to 80 mL of sterile water in a measuring cylinder. Bring the volume up to 100 mL with sterile water. Mix by carefully shaking the measuring cylinder until the solution appears homogeneous.
| Component | Stock solution (M) | Final concentration (mM) | Volume (mL) |
| NaCl | 5 | 114 | 2.3 |
| KCl | 3 | 5.29 | 0.176 |
| MgCl2 | 1.9 | 1 | 0.053 |
| CaCl2 | 1 | 2 | 0.2 |
| Glycine | 0.1 | 0.005 | 0.005 |
| Sucrose | 1.5 | 52 | 3.5 |
| Sodium pyruvate | 0.1 | 0.5 | 0.5 |
| HEPES | 1 | 10 | 1 |
| Glucose | 2.5 | 5 | 0.2 |
Table 2: Composition of imaging buffer. The indicated volumes are used for the preparation of 100 mL of imaging buffer.
- Solutions for stimulation and calibration
NOTE: Always prepare fresh stimulation solutions by adding stock solutions of indicated drugs to the imaging buffer just before the experiment. Solutions for stimulation and calibration will be added to the imaging chamber sequentially during an experiment (see sections 3-5). Depending on the type of experiment, different solutions are required to reach the same end concentration in the respective final volume in the imaging chamber.- Prepare 3x NMDA solution (90 µM; final concentration in the chamber: 30 µM) by adding 63 µL of a 10 mM NMDA stock to 6.937 mL of imaging buffer. Add 500 µL of the resulting solution to the chamber (final volume: 1.5 mL).
- Prepare 2x DA solution for steps 3 and 4 (1 mM; final concentration in the chamber: 0.5 mM) by adding 14 µL of a 0.5 M DA stock to 6.986 mL of imaging buffer. Add 1 mL to the chamber (final volume: 2 mL).
- Prepare 4x DA solution for step 5 (2 mM; final concentration in the chamber: 0.5 mM) by adding 28 µL of a 0.5 M DA stock to 6.972 mL of imaging buffer. Add 500 µL to the chamber (final volume: 2 mL).
- Prepare 1x DTT solution (5 mM; final concentration in the chamber: 5 mM) by adding 45 µL of 1 M DTT stock to 8955 µL of imaging buffer. Add 1 mL of this solution to the chamber after aspirating the imaging buffer (final volume: 1 mL).
2. Loading of cells with TMRE
NOTE: In this protocol, TMRE is used in non-quench mode15 at a final concentration of 20 nM. In general, the lowest possible concentration of TMRE that still provides sufficient signal intensity on the microscope of choice should be used. Due to uneven evaporation, the volume of medium in different wells can differ in long-term primary cultures. To ensure a consistent TMRE concentration in all wells, do not add TMRE directly to the wells. Instead, replace the medium in each well with the same amount of TMRE-containing medium. The protocol below is designed for primary neurons in 24-well plates containing ~1 mL of medium per well.
- Working in a tissue culture laminar flow hood, collect 500 µL of medium from each well into a single conical tube.
- Per well, add 0.5 µL of 20 µM TMRE stock into the conical tube (e.g., 12 µL for 24 wells).
- Carefully aspirate the remaining medium from the first well and replace it with 500 µL of TMRE-containing medium. Continue, well-by-well, with the remaining wells.
NOTE: Take care not to let the cells dry out and not to disturb the cells. - Return the cells to the incubator and wait for at least 60 min for dye equilibration.
NOTE: Loading time can be extended to several hours without adverse effects. - To ensure consistent TMRE concentrations and equilibration throughout the imaging experiment, make sure to include a final concentration of 20 nM TMRE in the imaging buffer and all stimulation solutions.
3. Optimization of scanning confocal microscope settings
NOTE: This step aims to find the best compromise between image quality and cell viability during live imaging. This section describes the optimization of settings for roGFP imaging. If multiparametric imaging is performed, similar optimization, including checking for a stable baseline without signs of bleaching or phototoxicity, needs to be performed for the additional indicators.
- Start the confocal microscope and load standard settings for GFP imaging (488 nm excitation, 505 – 550 nm emission).
- Set the detector to 12 bits or 16 bits.
NOTE: Usually, 8 bits are not sufficient for quantitative imaging. - Activate the sequential scan mode and add second sequence/track (405 nm excitation, 505 – 550 nm emission).
- For both channels, select a pseudocolor lookup table that indicates over- and under-exposed pixels (e.g., GLOW OU).
- Select an objective that is suitable for the object of interest.
NOTE: 10x-40x are suitable for single-cell analysis, 63x-100x are suitable for single-mitochondrion analysis. - Mount a coverslip with cells into the imaging chamber, add 1 mL of imaging buffer, and place the chamber on the microscope.
- Use the eyepiece and transmitted light to focus the cells.
NOTE: Do not use epifluorescence light to locate and focus cells. Even at low power, this will adversely affect the cells. - Record images with different pixel formats. Based on these images, select the lowest pixel number that gives an acceptable resolution of the structure of interest.
NOTE: Typically, 512 x 512 pixels work well for single-cell imaging with 20x and 40x objectives, and 1024 x 1024 or 2048 x2048 pixels typically work well for single-mitochondrion imaging with a 63x objective. - Record images with different pinhole sizes. Based on these images, select the largest pinhole size that gives an acceptable resolution of the structure of interest.
NOTE: Typically, 3-7 airy units work well. - Record images with different laser intensities.
- Adjust the detector gain and threshold accordingly. Based on these images, select the lowest laser intensity that gives acceptable signal intensity and signal-to-background ratio.
- To determine the signal-to-background ratio, measure the signal intensity in a region of interest (ROI) that contains cells or mitochondria (ROI1) and in an ROI without cells or mitochondria (ROI2). Then, divide the intensity of ROI1 by the intensity of ROI2.
NOTE: Aim for a signal-to-background ratio of >3 and signal intensities of individual ROIs of 200-1,000 for 405 nm excitation with 1-3% laser power and intensities of individual ROIs of 300-1,500 for 488 nm excitation with 1% laser power.
- To determine the signal-to-background ratio, measure the signal intensity in a region of interest (ROI) that contains cells or mitochondria (ROI1) and in an ROI without cells or mitochondria (ROI2). Then, divide the intensity of ROI1 by the intensity of ROI2.
- Adjust the detector gain and threshold accordingly. Based on these images, select the lowest laser intensity that gives acceptable signal intensity and signal-to-background ratio.
- Record images with different scan speeds and number of frame averages. Record 4-5 images for each combination of settings. Based on these image series, select the highest speed and lowest average settings that give acceptable image noise and image-to-image variability.
NOTE: A scan speed of 600 Hz and 1-2 frames for averaging work well in most cases. - Using a new coverslip, record a time-lapse series with the optimized settings.
NOTE: The duration and image interval of the series should resemble those of the planned experiments. - At the end of the time-lapse series, add 1 mL of 2x DA solution to the recording chamber. Image for additional 2 min.
- Aspirate the imaging buffer using a peristaltic pump or handheld pipette. Add 1 mL of 1x DTT solution. Image for additional 5 min.
- Analyze the time-lapse experiment (see section 5).
- Verify that none of the two channels gets over- or under-exposed during DA- and DTT-treatment with the optimized settings.
- Ensure that none of the two channels shows considerable bleaching during the time-lapse recording; aim for <2% loss of intensity between the first and last images.
- Verify that the 405:488 ratio does not change considerably during imaging.
- Repeat the whole procedure in an iterative manner, using several coverslips, until settings that consistently provide acceptable results have been defined.
4. Assessment of basal redox status
- Start the microscope and load the optimized settings from section 3.
- Set frame average to 3-5.
- Mount a coverslip with cells into the imaging chamber, add 1 mL of imaging buffer, and place the chamber on the microscope.
- Use the eyepiece and transmitted light to focus the cells.
NOTE: Do not use epifluorescence light to locate and focus cells. Even at low power, this will adversely affect the cells. - Switch to scanning mode and use the 488 nm channel in live view to focus and locate cells for imaging.
- Use the multipoint function to select 3-5 fields of view on the coverslip.
- Record a baseline image.
- Add 1 mL of 2x DA solution to the chamber.
- After 1, 2, and 3 min, use live view to confirm/adjust the focus and then record an image.
NOTE: Cells are typically fully oxidized after 2 min. - Replace the buffer in the imaging chamber with 1 mL of 1x DTT solution.
- After 3 and 5 min, use live view to confirm/adjust the focus and then record an image.
NOTE: Cells are typically fully reduced after 4-5 min.
5. Live imaging of acute treatments
NOTE: The protocol below describes imaging of the mitochondrial redox response to NMDA treatment. Image intervals and duration of the experiment might need to be adjusted for other treatments.
- Start the microscope and load the optimized settings from section 3.
- Set the time-lapse interval to 30 s and duration to 25 min.
- Mount a coverslip with cells into the imaging chamber, add 1 mL of imaging buffer, and place the chamber on the microscope.
NOTE: To avoid thermal focus drift, leave the cells on the microscope stage for 10-15 min before starting time-lapse imaging. - Use the eyepiece and transmitted light to focus the cells.
NOTE: Do not use epifluorescence light to locate and focus cells. Even at low power, this will adversely affect the cells. - Switch to the scanning mode and use the 488 nm channel in live view to focus and locate cells for imaging.
- Optional: To increase the number of recorded cells per run, use the multipoint function to image 2-3 fields of view per coverslip.
- Start the time-lapse acquisition and record 5 images as 2 min baseline recording.
- Add 500 µL of 3x NMDA solution to the chamber (final concentration 30 µM) and record additional 20 images as a 10 min NMDA response.
NOTE: Neurons are very sensitive to changes in osmolarity. Therefore, make sure to minimize evaporation of the imaging buffer. For longer treatments, the imaging chamber should be covered with a lid. - Add 500 µL of 4x DA solution to the chamber and record 6 more images (3 min maximum calibration).
- Aspirate the buffer from the imaging chamber and replace it with 1 mL of 1x DTT solution. Record 10 more images (5 min minimum calibration).
- End the recording and save the image series.
6. Data analysis
- Data import and image preprocessing in FIJI
- Use the Bio-Formats Importer to open a group of images from step 4 or an image file from step 5. Click on Plugins | Bio-Formats | Bio-Formats Importer. In the dialog box, use View stack with: Hyperstack, set Color mode: default, select Autoscale, and do not split into separate windows.
NOTE: Autoscale optimizes the display of the data on the computer screen. It does not change pixel intensities. - If individual images from step 4 were opened, click on Image | Stacks | Tools | Concatenate to merge them into a single-image stack.
- If there is XY-drift during the image series, click on Plugins | StackReg to register the images. In the dialog box, select Rigid Body or Translation.
- Change the image format to 32 bit by clicking on Image | Type | 32-bit.
- Split the color channels into separate windows by clicking on Image | Color | Split Channels.
- Select channel 1 (405 nm) and adjust the threshold to select the mitochondria for analysis by clicking on Image | Adjust | Threshold. In the dialog box, select Default, Red, Dark background, and Stack histogram and wait for the selected pixels to appear red. Click Apply. Select Set Background Pixels to NaN and Process all images.
NOTE: To avoid potential observer bias, automated threshold determination should be used. FIJI offers several automated methods (such as Default, Huang, Intermodes, Otsu) that can be selected from a dropdown menu in the threshold dialog box. Typically, the Default method gives a good result. It is recommended to compare several methods during the first analysis to find the best thresholding method for the given set of images. Once a method has been chosen, it needs to be applied to all images. - Repeat step 6.1.6 for channel 2 (488 nm).
- Create a ratio image to visualize the 405:488 nm ratio by clicking on Process | Image calculator. In the dialog box, select Image 1: channel 1, Operation: Divide, Image 2: channel 2, Create new window, Process all images.
- Change the lookup table of the ratio image to pseudocolor. For example, to change to Fire, click on Image | Lookup Tables | Fire.
- Use the Bio-Formats Importer to open a group of images from step 4 or an image file from step 5. Click on Plugins | Bio-Formats | Bio-Formats Importer. In the dialog box, use View stack with: Hyperstack, set Color mode: default, select Autoscale, and do not split into separate windows.
- Image analysis
- On the ratio image, draw ROIs around individual cells or mitochondria. After drawing each ROI, add it to the ROI Manager. Analyze | Tools | ROI Manager | Add. (keyboard shortcut: 'T') Select Show All.
NOTE: Because background pixels have been set to 'not a number' (NaN) in steps 6.1.6 and 6.1.7, they will not affect the result of the measurement. Therefore, it is acceptable to include some background pixels in the ROI. - Measure the 405:488 ratios of individual cells by clicking on ROI Manager | ctrl+A to select all ROIs | More | Multi Measure. In the dialog box, select Measure all slices and One row per slice.
- Export the measurements to spreadsheet software.
- Select the 405 nm image. Measure the intensities of all ROIs as in step 6.2.2. using the ROIs that are stored in the ROI manager.
- Export the measurements to spreadsheet software.
- Select the 488 nm image. Measure intensities of all ROIs as in step 6.2.2. using the ROIs that are stored in the ROI manager.
- Export the measurements to spreadsheet software.
- Save ROIs for future reference by clicking on ROI Manager | ctrl+A to select all ROIs | More | Save.
- Recommended: Generate intensity vs. time plots of the 405 and 488 nm traces. Verify that there is no marked bleaching in either of the channels (signal intensity at the end of the imaging series should be ≥98% of the first image) and that the two traces move into opposite directions during sensor responses (e.g., the 405 nm trace should increase during oxidation while the 488 nm trace should decrease).
- On the ratio image, draw ROIs around individual cells or mitochondria. After drawing each ROI, add it to the ROI Manager. Analyze | Tools | ROI Manager | Add. (keyboard shortcut: 'T') Select Show All.
- Data normalization
- For each ROI from the ratio image, determine the maximum value during DA treatment (Rmax) and the minimum value during DTT treatment (Rmin).
- Calculate the normalized ratio as follows:
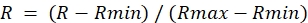
NOTE: This will set the maximum ratio to 1.0 and the minimum ratio to 0.
- Analysis of mitochondrial morphology
- To obtain measurements of mitochondrial morphology in parallel to roGFP intensities in step 6.2.6, go to Analyze | Set Measurements and check Shape descriptors and Fit ellipse.
NOTE: In addition to mean intensity, the measurements in the results window will include the length of the major axis (Major), the length of the minor axis (Minor), the aspect ratio (AR; major axis divided by minor axis; round mitochondria have an AR ~1, elongated mitochondria have a greater AR), as well as measurements of circularity (Circ.) and roundness (Round).
- To obtain measurements of mitochondrial morphology in parallel to roGFP intensities in step 6.2.6, go to Analyze | Set Measurements and check Shape descriptors and Fit ellipse.
Quantification of differences in steady-state mitochondrial redox state after growth factor withdrawal
To demonstrate the quantification of steady-state differences in mitochondrial redox state, primary neurons grown in standard medium were compared to neurons cultured without growth factors for 48 h before imaging. Growth factor withdrawal results in apoptotic neuronal cell death after 72 h16. Cells were imaged after 48 h to test if this is preceded by changes in mitochondrial redox state. Primary rat cortical neurons grown on poly-L-ornithine-coated coverslips were infected with rAAV-mito-Grx1-roGFP2 on days in vitro 6 (DIV6) and were imaged on DIV12. Live imaging was performed at room temperature according to section 4 of this protocol on an inverted laser scanning confocal microscope equipped with a 40x/1.10 water immersion objective. Confocal settings were pinhole 7 airy units, pixel size 568.7 nm (512 x 512 pixels), scan speed 600 Hz, laser power 405 nm 3%, laser power 488 nm 1%, emission bandwidth 505-550 nm, and frame average 4. There was no major difference between the raw 405:488 nm ratios of the two conditions (Figure 3B). After data normalization, a subset of cells with an increased 405:488 nm ratio was detectable in the growth factor withdrawal group (Figure 3C). This indicates that mitochondrial redox changes might precede neuronal cell death, and it underscores the relevance of max/min data normalization for the comparison of basal redox states between groups.
Dynamic changes in mitochondrial redox state upon treatment of neurons with NMDA
The NMDA-type glutamate receptor (NMDAR) plays a central role in neuronal plasticity but can also mediate neuronal damage and cell death. Pathological activation of the NMDAR leads to several adverse effects on mitochondria that include matrix calcium overload, mitochondrial oxidation and fragmentation, and mitochondrial permeability transition. In a previous study, the above-described protocol was used to investigate a causal relationship between NMDA-induced mitochondrial calcium overload and mitochondrial oxidation13. Primary rat hippocampal neurons grown on poly-D-lysine/laminin-coated coverslips were infected with rAAV-mito-Grx1-roGFP2 on DIV4 and imaged on DIV12. Live imaging was performed at 37 °C on an inverted spinning disc confocal microscope equipped with a 20x/0.75 multi immersion objective (water immersion was used) and an on-stage incubation system. Mito-Grx1-roGFP2 was sequentially excited every 20 s using the 405 nm and 488 nm laser lines, and emission was collected with a 527/55 nm emission filter for both excitation wavelengths. Treatment of neurons with 30 µM NMDA caused oxidation of mitochondria within a few minutes (Figure 4A,B). Notably, NMDA-induced mitochondrial acidosis caused significant quenching of roGFP2 fluorescence, in line with its well-known pH-sensitivity8. To confirm that this pH-dependent quenching did not affect the 405:488 ratio9, mitochondria were fully oxidized by DA before the addition of NMDA in a control experiment. Pretreatment with DA precludes any further oxidation of mitochondria by NMDA and, accordingly, the 405:488 ratio did not change in this experiment despite a considerable quenching of roGFP2 fluorescence intensity (Figure 4C).
Multiparametric analysis of NMDA-induced changes of dendritic mitochondria
To assess the temporal sequence of NMDA-induced changes in mitochondrial morphology, membrane potential, and redox state, parallel imaging of TMRE- and mito-Grx1-roGFP2 fluorescence was performed at high spatial and temporal resolution. Primary rat cortical neurons grown on poly-L-ornithine-coated coverslips were infected with rAAV-mito-Grx1-roGFP2 on DIV6 and imaged on DIV12. Live imaging was performed at room temperature on an inverted confocal laser scanning microscope using a 63x/1.40 oil immersion objective, a scan speed of 600 Hz, a pixel size of 90.2 nm (1024 x 1024 pixels at 2x scan zoom), a pinhole size of 3 airy units, and a frame average of 2. Every 30 s, three images were recorded in sequential mode: 405 nm excitation/505-550 nm emission; 488 nm excitation/505-550 nm emission; 552 nm excitation/560-600 nm emission. Treatment of neurons with 60 µM NMDA resulted in a loss of TMRE signal and an increase in the 405:488 nm roGFP ratio, followed by some delayed rounding up of mitochondria (Figure 5).
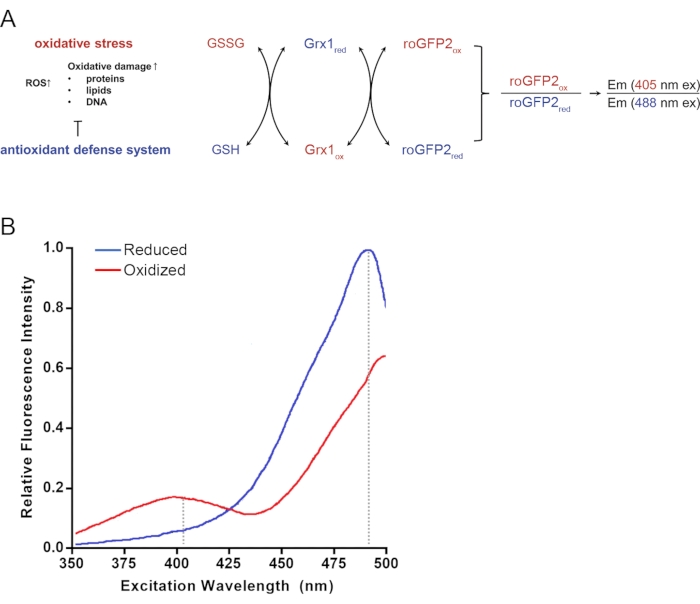
Figure 1: Schematic representation of Grx1-roGFP2 function and roGFP2 excitation spectra. (A) Oxidative stress and the action of antioxidant defense systems oxidize the cellular glutathione pool. Grx1 in the Grx1-roGPF2 fusion protein promotes the rapid equilibration of the roGFP2 redox state with the redox state of the glutathione pool. The redox status of the roGFP2 pool can be assessed by monitoring the ratio of GFP-fluorescence emission at 510 nm after excitation at 405 nm and 488 nm. Reduced species are shown in blue; oxidized species are shown in red. (B) Excitation spectra of fully reduced (blue) and oxidized (red) roGFP2. Upon oxidation of roGFP2, fluorescence emission at 400 nm excitation increases, whereas emission at 490 nm excitation decreases. This figure has been modified from a previous publication13. Excitation spectra in B were drawn based on Figure 1B from 8. Dotted lines in B indicate the wavelengths of commonly used 405 nm and 488 nm laser lines. Abbreviations: GSH = glutathione; GSSG = oxidized glutathione; Grx = glutaredoxin; roGFP = redox-sensitive green fluorescent protein variant. Please click here to view a larger version of this figure.
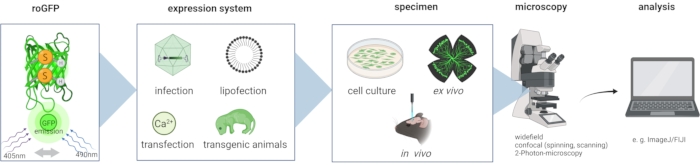
Figure 2: Workflow of the method. Expression of the excitation-ratiometric redox-sensitive fluorescent protein roGFP2 in neurons can be achieved through several methods that include transfection, lipofection, viral gene transfer, and transgenic animals. The sensor can be used to study the neuronal redox state in cultured primary neurons, ex vivo tissue explants, and intact animals. roGFP2 imaging can be performed on a variety of microscopes that include widefield fluorescent microscopes, confocal microscopes, and 2-photon microscopes. Analysis of roGFP2 imaging data can be performed with the freely available software ImageJ/FIJI. Abbreviation: roGFP = redox-sensitive green fluorescent protein variant. Please click here to view a larger version of this figure.
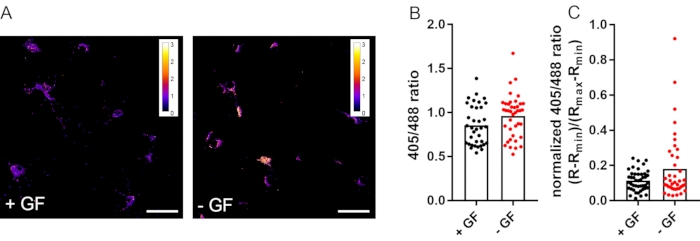
Figure 3: Growth factor withdrawal causes oxidation of neuronal mitochondria. (A) Representative 405:488 nm ratio images of neurons cultured in the presence (+ GF) or absence (- GF) of growth factors for 48 h before imaging. Color-coded scales represent non-normalized 405:488 nm ratios (lower ratios correspond to a reduced state; higher ratios correspond to an oxidized state). Scale bars = 50 µm. (B) Quantification of the 405:488 nm ratio in individual neurons. (C) Max/min calibrated 405:488 nm ratio of individual neurons. Round symbols represent single cells; bar represents mean. N = 40-44 cells from 3 coverslips from one preparation. Abbreviation: GF = growth factor. Please click here to view a larger version of this figure.
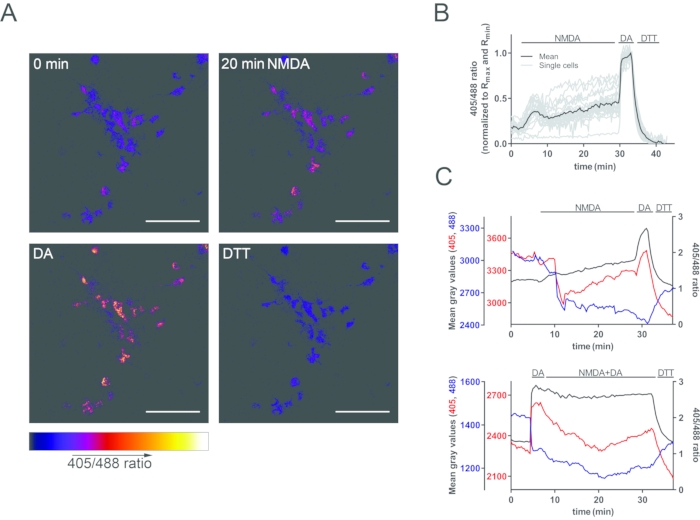
Figure 4: NMDA-induced oxidation of neuronal mitochondria. (A) Representative 405:488 nm ratio images before and after treatment with 30 µM NMDA and after max/min calibration with DA and DTT. At this magnification, roGFP signal is mostly detected in the soma and proximal dendrites. The color-coded scale represents non-normalized 405:488 nm ratios (lower ratios correspond to a reduced state; higher ratios correspond to an oxidized state). Scale bars = 50 µm. (B) Quantification of the imaging run shown in A. NMDA induces a rapid and sustained mitochondrial oxidation that can be calibrated using DA and DTT. (C) NMDA-induced mitochondrial acidosis causes a drop of GFP fluorescence upon both 405 nm and 488 nm excitation (upper panel). To isolate the pH-driven effect and confirm that the 405:488 nm ratio is pH-insensitive, neurons were first maximally oxidized using DA and subsequently challenged with NMDA in the presence of DA (lower panel). Under these conditions, NMDA still causes mitochondrial acidosis but no further mitochondrial oxidation. Accordingly, although both the 405 nm and 488 nm trace show a pH-driven drop in fluorescence intensity, the 405:488 nm ratio remains stable. This figure is modified from 13. Abbreviations: GFP = green fluorescent protein; roGFP = redox-sensitive green fluorescent protein variant; NMDA = N-methyl-D-aspartate; DA = diamide; DTT = dithiothreitol. Please click here to view a larger version of this figure.
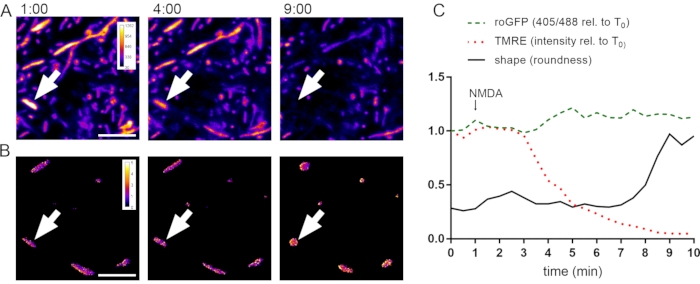
Figure 5: NMDA-induced changes in membrane potential, redox state, and morphology of dendritic mitochondria. (A) Three high-magnification images from a time-lapse experiment, acquired at t = 1, 4, and 9 min, showing dendritic and axonal mitochondria. Colorization represents TMRE intensity (see calibration bar). (B) 405:488 nm roGFP ratio images from the same time-lapse experiment as in (A) at t = 1, 4, and 9 min. Note that due to limited AAV infection efficiency, only a subset of neurons expresses mito-Grx1-roGFP2, and therefore, not all TMRE-positive mitochondria are roGFP-positive. The color-coded calibration bar represents non-normalized 405:488 nm ratios (lower ratios correspond to a reduced state; higher ratios correspond to an oxidized state). (C) Quantification of TMRE intensity, roGFP 405:488 nm ratio, and roundness of a single mitochondrion (indicated by arrows in A, B). After 1 min of baseline recording, 60 µM NMDA was added to the bath solution. Scale bars = 5 µm. The y-axis in C depicts the 405:488 nm roGFP ratio relative to baseline T0 (green dashed line), the TMRE fluorescence intensity relative to baseline T0 (red dotted line), and the FIJI/ImageJ shape descriptor "roundness" (black solid line). Abbreviations: GFP = roGFP = redox-sensitive green fluorescent protein variant; mito-Grx1-roGFP2 = Glutaredoxin-1 fused to N-terminus of roGFP; AAV = adeno-associated virus; TMRE = tetramethylrhodamine, ethyl ester; NMDA = N-methyl-D-aspartate. Please click here to view a larger version of this figure.
