Experiments were performed on isolated nerve-muscle preparations of levator auris longus (m. LAL) from the Mice BALB/C (20-23 g, 2-3 months old)41. The experimental procedures were performed in accordance with the guidelines for the use of laboratory animals of the Kazan Federal University and the Kazan Medical University, in compliance with the NIH Guide for the Care and Use of Laboratory Animals. The experimental protocol met the requirements of the European Communities Council Directive 86/609/EEC and was approved by the Ethical Committee of the Kazan Medical University.
1. Preparation of the Ringer's and Filing solutions
- Prepare the Ringer's solution for mammalian muscle by mixing the following ingredients: NaCl (137 mM), KCl (5 mM), CaCl2 (2 mM), MgCl2 (1 mM), NaH2PO4 (1 mM), NaHCO3 (11.9 mM), and glucose (11 mM). Bubble through the solution with 95% O2 and 5% CO2 and adjust its pH to 7.2-7.4 by adding HCl/NaOH if necessary.
- Prepare the dye loading solution.
- Prepare HEPES (10 mM) solution with pH in the 7.2-7.4 range. 500 µg of commercial dye comes in a 500 µL vial. Dissolve the dye in 14 µL of the HEPES solution to obtain a dye concentration of 30 mM. Shake well and centrifuge until totally dissolved.
- Dilute the solution of Ca2+ indicator with HEPES solution down to 1 mM concentration. Keep it in a freezer (-20 °C) and avoid exposure to light.
2. Dye loading procedure
NOTE: The dye loading procedure is performed according to the protocol for loading through the nerve stump, adapted from the protocols previously published19,42,43,44,45,46.
- Dissect LAL muscle according to the dissection procedure for this preparation as described in the previously published protocols47,48.
- Fix the tissue slightly stretched (no more than 30% from initial length) in the elastomer-coated Petri dish with fine stainless-steel pins and add Ringer's solution until the muscle is fully covered.
NOTE: The Petri dish was pre-filled with elastomer according to the manufacturer's instructions (see Table of Materials).
- Fix the tissue slightly stretched (no more than 30% from initial length) in the elastomer-coated Petri dish with fine stainless-steel pins and add Ringer's solution until the muscle is fully covered.
- Prepare the Filling Pipette
- Using a micropipette puller (see Table of Materials), prepare a micropipette with a fine tip which is as sharp as possible for the intracellular recordings. Use capillaries without internal filaments (1.5 mm in outer diameter and 0.86 or 1.10 mm in inner diameter).
- Break off the micropipette tip after scoringthe taper with an abrasive, leaving the tip open to about 100 µm in diameter. Fire-polish the tip down to limit when the internal diameter shrinks from >80 µm to 12-13 µm. Attach a silicone tube to one side of the Filling Pipette and a syringe (without a needle) to the other side.
- Under a stereomicroscope, find the place where the nerve trunk turns into separate nerve branches. Place the Filling Pipette with the mounted tube and the syringe on the Petri dish using wax. Move the pipette tip until it stands above the nerve.
- With fine scissors, cut the nerve close to the muscle fiber, leaving a small piece of the nerve stump about 1 mm long. Gently aspirate the nerve stump together with some Ringer's solution, without pinching it, into the tip of the Filling Pipette. Remove the silicone tube from the Filling Pipette.
- Draw some amount of the dye loading solution (~0.3 µL) using a syringe with a long filament. This volume corresponds to approximately 3 cm of the filament.
NOTE: Initially, it is necessary to make a filament from a pipette tip with a volume of 10 µL by pulling on the fire using an alcohol lamp or a gas burner. - Gently insert the filament tip with loading solution into the Filling Pipette. Release the mixture directly onto the nerve stump. Incubate the preparation at room temperature in dark for 30 min.
- After that, rinse the preparation with fresh Ringer's solution and incubate at 25 °C for up to 2 h in a glass beaker with 50 mL (or more) Ringer's solution (preparation must be covered with the solution). During this time, the dye will reach the synapses.
3. Video capture with confocal microscopy
NOTE: Registration of calcium transients is performed with a laser scanning confocal microscope (LSCM) (see Table of Materials). To register fast calcium transients, an original protocol that permitted recordings of signals with a sufficient spatial and temporal resolution was used. The method has been described thoroughly in the publication by Arkhipov et al37. The microscope was equipped with a 20x water immersion objective (1.00 NA). The 488 nm laser line was attenuated to 10% intensity and emission fluorescence was collected from 503 to 558 nm.
- Mount the preparation into the silicon elastomer-coated experimental chamber and fix it, slightly stretched, with a set of steel micro-needles. Rinse the preparation extensively with Ringer's solution.
NOTE: A simple custom-made perfusion experimental chamber made of organic glass with the bottom of the chamber covered with an elastomer (prepared in accordance with the manufacturer's instructions; see Table of Materials) was used. The chamber has a solution supply tube. The solution is pumped out via a syringe needle, mounted on a magnetic holder (see Table of Materials). As an experimental chamber, a Petri dish could be used (like the one used for incubation of the preparation) but with attached supply and suction tubes. - Install suction electrode which will be used to stimulate the nerve.
NOTE: Construction of the electrode is similar to what was published in the 2015 paper by Kazakov et al49. Place and fix the electrode by waxing beside the bath. Move the tip close to the nerve stump and aspirate it into the electrode. - Mount the preparation chamber onto the microscope stage and place the inlet and outlet fittings into the chamber.
- To perfuse the preparation, use a simple gravity-flow-driven system. Turn on the perfusion suction pump to remove the excess solution.
- Plug the stimulating suction electrode into an electric stimulator and ensure that muscular contractions occur after stimuli. See section 3.9-3.12 for stimulation conditions and recording.
- Fill up the perfusion system with the Ringer's solution with d-tubocurarine (10 µM).
NOTE: This solution helps to prevent muscular contractions. D-tubocurarine or alpha-bungarotoxin-specific blockers of nicotinic acetylcholine receptors on the postsynaptic membrane would completely or partially block muscle contractions50. Also, for preventing muscular contractions, specific blockers of postsynaptic sodium channels such as µ-conotoxin GIIIB could be used51. - Switch on the perfusion suction pump and start perfusion of the preparation with the Ringer's solution containing d-tubocurarine.
- Set imaging parameters in the LSCM software as follows.
- In the LSCM software (LAS AF; see Table of Materials), choose Electrophysiology.
NOTE: In this mode, when an image is captured at the time point, a synchronizing pulse is sent to the stimulator with the help of the trigger box. This elicits action potential generation in the preparation (Figure 1; stimulator unit). - Select Acquisition Mode. For triggering the stimulator using the microscope sync pulse, in the Job menu settings, select the Trigger settings. Set the Trigger Out On Frame field to the out1 channel.
- Use the following settings: Scanning Mode: XYT, Frequency of Scanning: 1400 Hz, Zoom Factor: 6.1, Pinhole: fully open. Ensure that sequential trans-passing Bidirectional X mode is on.
- Set minimum time to form a frame at 52 ms and frames to be collected in a raw video at 20 frames.
NOTE: These settings permit image capturing with a resolution of 128 x 128 pixels while taking a single frame every 52 ms. - Set excitation wavelength of the argon laser at 488 nm with 8% of output power.
- In the LSCM software (LAS AF; see Table of Materials), choose Electrophysiology.
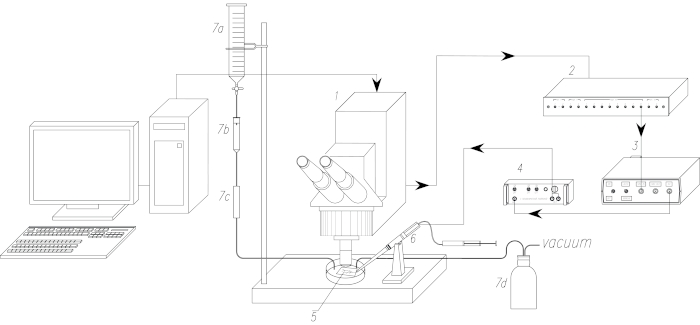
Figure 1: The schematic of the experimental setup. 1. Laser Scanning Confocal Microscope (LSCM). 2. Synchronization module of LSCM (trigger box). 3. Stimulator. 4. Isolation unit. 5. The biological sample. 6. Suction electrode for electrical stimulation of nerve. 7. Perfusion systems (7a: perfusate reservoir, 7b: dropper, 7c: flow regulator, 7d: vacuum flask). Arrows point to the direction of propagation of synchronizing pulse. Please click here to view a larger version of this figure.
- Press the Live Mode button to switch to Live mode, which helps to get a preview of nerve terminals loaded with the dye.
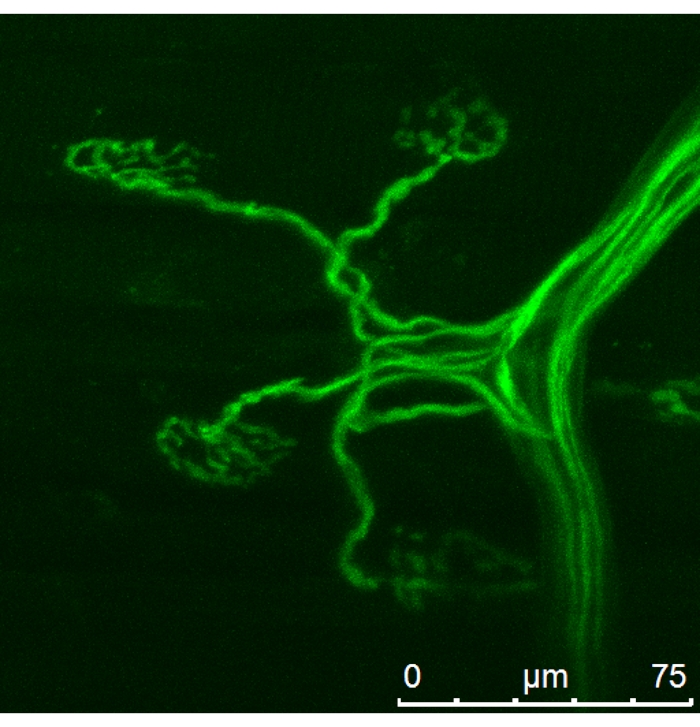
Figure 2: Mouse nerve and terminals loaded with the Ca2+ indicator. Please click here to view a larger version of this figure.
- Stimulation unit
NOTE: In this work, the stimulator described in the article by Land et al.52 was used. This device allows for setting temporal parameters of stimulation via the MatLab software.- Create a new file, paste the code from the above-mentioned article to the MatLab code window, and save the file. Click on Run, so a window with stimulation parameters appears. Set the delay time and duration of the stimulus.
NOTE: The delay determines the temporal resolution of the reconstituted fluorescent signal. The electric pulse of 0.2 ms duration is delayed, and then sent to the isolation unit. The latter forms the amplitude and polarity of the stimulating pulse and electrically isolates the biological object from the recording equipment. - To stimulate the nerve, select supramaximal amplitude of the stimulating impulse (25%-50% greater than the maximum stimulation intensity necessary to activate all the nerve fibers).
NOTE: The presented method is based on a special algorithm for recordings of single fast fluorescent signals using LSCM with the minimized sweep. At each step of the developed algorithm, the recorded fluorescent signal is shifted from the previous one by a time interval that is shorter than the microscope sweep. The value of time shifts determines the temporal resolution of the required signal. The number of steps (shifts) in the algorithm depends on the required temporal resolution and original temporal resolution. With this method of registration, the stimulation of the preparation is carried out with a frequency of 0.25 Hz.
- Create a new file, paste the code from the above-mentioned article to the MatLab code window, and save the file. Click on Run, so a window with stimulation parameters appears. Set the delay time and duration of the stimulus.
- In the Live mode, search for the ROI and obtain the best focus. Run the data acquisition software.
- Shift the delay on the stimulator by 2 ms less relative to the previous value and run the data acquisition software.
- Repeat step 3.11 26 times to acquire 26 sequences, with each sequence shifted by 2 ms from the previous one.
4. Video processing
NOTE: A series of video images acquired by the confocal microscope is exported in the TIFF format with the free software LAS X (see Table of Materials). This series was divided into frames and exported to a folder. For generating the image sequence with higher time resolution, the ImageJ software, which has an open initial code for the analysis and processing of the data, was used. The algorithm of signals processing is represented schematically in Figure 3.
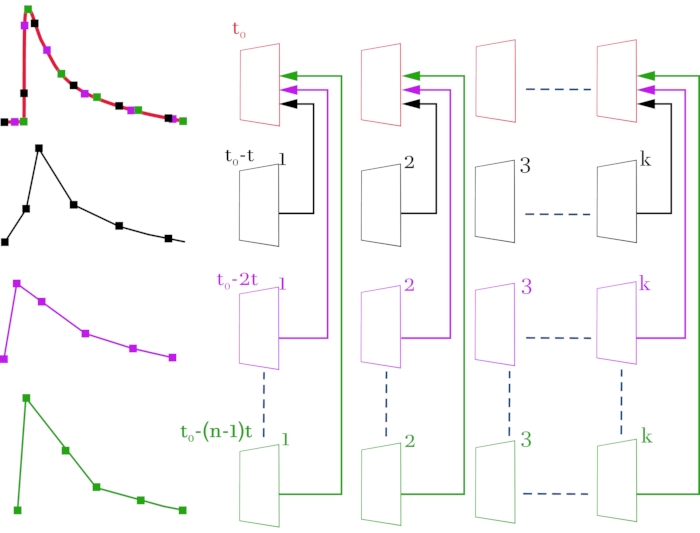
Figure 3: Scheme for compiling a high-resolution video file (2 ms on frame) from original video files with a low temporal resolution (52 ms on frame). The original video files and the corresponding signals are colored in black, magenta, and green. The compiled video file and the resulting signal are colored red. The scheme on the right, line by line, shows the video images obtained with a confocal microscope. On the left, the corresponding signals of fluorescence change from the selected ROI. The topmost line is formed frame by frame from the received frames according to the scheme. The result is a video image consisting of the entire array of frames so that there is a delay time of 2 ms between frames instead of 52 ms. Each line corresponds to an offset of the stimulation signal by (n – 1) * t, where t is time shift (2 ms), and n is the number of shift iterations. k denotes the number of frames in the original video files (lines 2-4) and depends on the duration of the recorded signal. In this case, to register a signal with a duration of 1 s, it is necessary to select k = 20 (52 ms * 20 = 1040 ms). t0 is the required delay before stimulation. To calculate the number of shift iterations n, the initial temporal resolution between frames (52 ms) must be divided by the required temporal resolution (2 ms). In this case, n = 26, which corresponds to 26 registered sweeps. As a result of the performed manipulations, a video image consisting of n * k = 520 frames is obtained. Please click here to view a larger version of this figure.
- Run LAS X software. Open the project which was created during performing experiment. Click on Export, and then on Save As to save frames in .tiff format in the destination folder.
- Run the ImageJ software. Click on File > Import > Image Sequence.
- In the Open Image Sequence window, choose the destination folder and open the first frame.
- In the Sequence Options window, in the Starting Image field, set the frame number to 1 for the first frame. In the Increment field, set the value equal to the number of frames in the initial signal recording (20 for the present case) and click on OK.
- To save the generated file of stitched first frames in a separate folder, click on File > Save > Folder.
- Repeat steps 4.3-4.5 for the next 19 frames. In the Sequence Options window, set the corresponding frame number in the Starting Image field.
- To generate the full high-time-resolution video, stitch all the frames together. To do this, click on File > Import > Image Sequence and select 1 in the Starting Image and Increment fields. The result will be the final video with increased temporal resolution. Save the file in .tiff or any other suitable format.
5. Video analysis
NOTE: In ImageJ, select ROI and background. Subtract background from ROI. Data is represented as the ratio, (ΔF / F0 – 1) * 100%, where F0 is the intensity of fluorescence at rest and ΔF is the intensity of fluorescence during stimulation.
- Click on Image > Stacks > Tools > Stack Sorter. Then, click on Analysis > Tools > ROI Manager.
- Drag and drop the .tiff file saved in step 4.7 into the ImageJ window. Expand the image for a better view. To improve image visualization, click on Image > Adjust > Brightness/Contrast > Auto. This step will not affect the data.
- Set the background close to the nerve terminal by drawing ROI. Add it to the ROI manager. Calculate the background by clicking on More > Multi Measure. Copy mean values, paste to the Spreadsheet program, and calculate the average.
- Subtract the calculated average value from the stacks by clicking on Process > Main > Subtract. Enter the value.
- Draw ROI around a nerve terminal via a polygon line. Add it to the ROI manager.
- Measure the intensity of the nerve terminal: Click on More > Multi Measure. Copy mean values and paste them to the Spreadsheet program.
- Calculate the average offset of signals.
NOTE: Use the corresponding points depending on the delay time before stimulation. This step establishes the F0 value that will be used in subsequent calculations. - Divide the signal values by the average offset value.
NOTE: After this step, the signal does not contain the contribution of the background and raw fluorescence to the amplitude values for the selected ROI. - Subtract 1 from values obtained in step 5.8, and then multiply by 100%.
- Plot a graph of Ca2+– transient and calculate the amplitude.
After loading the preparation with dye according to the presented technique, most of the synapses located close to the nerve stump had a sufficient level of fluorescence (see Figure 2). After loading preparation with the dye andapplying the described method of registration and image processing, calcium transients with the desired spatial and temporal resolution were obtained (see Figure 4). The calcium transient has been recovered by the proposed method (see Figure 3).
Amplitude and time parameters of the recovered signals were also analyzed. Average data are presented in Table 1.
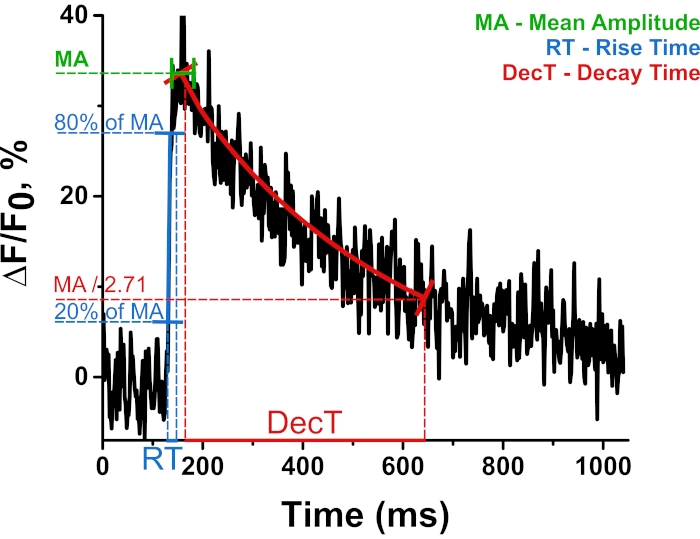
Figure 4: Representative trace of calcium signal from one experiment. Some important parameters of signal, such as mean amplitude (MA), rise time (RT), and decay time (DecT) and its projections on axes are indicated. MA is calculated by averaging points at the peak, colored in green. RT is the time taken for the amplitude to rise from 20% to 80%, which is calculated as the difference between projections on the x-axis colored in blue. DecT is the time over which the amplitude decreases by e times, which is calculated as the difference between projections on the x-axis colored in red. Please click here to view a larger version of this figure.
| Peak ΔF/F (%) | Rise time 20%-80% (ms) | τ (ms) |
| 27.0±4.6 (n=5) | 6.8±0.48 (n=5) | 456±53(n=5) |
Table 1: The averaged parameters of the Ca2+ transient. Data are presented as mean ± s.e.m., and n is the number of measurements in the distinct nerve-muscle junctions. Peak ΔF/F is the mean amplitude of ΔF/F.
Calcium transient analysis makes it possible to assess the amplitude-dynamic characteristics of changes in the presynaptic calcium level in the nerve ending during the action potential11. The change in the amplitude of the calcium transient correlates well with the change in the quantal content53. Calcium transient amplitude analysis is commonly used to study the effect of physiologically active compounds associated with modulation of presynaptic calcium levels on synaptic transmission54,55. The time course of the calcium transient reflects the kinetics of calcium binding with the dye and its dissociation23,56. It is obvious when using dyes with different affinity for calcium23,56. Although the temporal parameters of the calcium transient reflect the kinetics of the calcium sensitive dye, and do not represent the kinetics of free calcium in the nerve terminal, mathematical modeling methods based on experimental data can restore the behavior of free calcium in the cell and calculate the concentration of calcium buffers23.
